

The brain is magnificent!
The intricacies of athletic performance, the brilliance of mathematical theory, the artistry of a musical composer, the contemplative reflection of faith, the very breath we take… all of this appears to be based on a series of complex biochemical interactions in the organ that we call the brain. These interactions underlie our life, embodied as humans.
In this page, we will present an overview of some of those complex interactions and their relationship to vitamin K. We will explain some of the key elements of the brain, how the elements interact via signaling systems, and their impact on aging. Throughout, we will review the research on how vitamin K, and select vitamin K dependent proteins are essential in supporting brain function.... as it relates to Alzheimer’s Disease.
The Brain
The brain is the eminent organ of the human body. It controls thought, memory, emotion, speech, touch, motor skills, vision, temperature, hunger and every process that regulates our body, including many unconscious functions of the body such as the respiratory process and heart rate (Ecker et al, 2015). It is the palpable essence of the mind and soul. All of this happens via neurons, which serve as the primary center for initiation, coordination, interpretation, and integration of most of the nerve messages that are implemented in the brain (Hussain et al, 2013).
The brain can be divided into four main lobes: temporal, parietal, occipital and frontal, which are highly specialized. The temporal lobe is involved in laying down long-term memories, processing sensory input, and assigning emotional meaning. Within the temporal lobe is the hippocampus, and the entorhinal cortex whose functions include a widespread network hub for memory, navigation, and the perception of time. Dementia often affects the temporal lobe first. Later, damage to the frontal lobe leads to difficulties with cognitive function, judgment and behavior. When damage spreads to the parietal lobe, then language processing becomes impaired, among other things.
The nervous system is made up of the central nervous system and the peripheral nervous system. Together, the brain and the spinal cord that extends from it, make up the central nervous system or the CNS. It is referred to as central because it combines information from the entire body and coordinates activity across the whole organism. The CNS consumers 20% of the body’s oxygen and is highly susceptible to oxidative damage (Liguori et al, 2021). The nerves that go through the whole body make up the peripheral nervous system. Nerves provide a common pathway for electrochemical communication to and from tissues throughout the body.
There are many components of the nervous system. Some of the key components are discussed below.
The brain is composed of two types of cells: glial cells and neurons. Glial cells provide structural and metabolic support for the brain. Neurons are excitable cells which chemically transmit electrical signals through connections called synapses.
Neurons
Neurons are the fundamental units of the brain and nervous system. They use electrical impulses and chemical signals to transmit information within the brain, and between the brain and the rest of the nervous system. Research suggests the human brain consists of an estimated 100 billion neurons. Each neuron forms connections to other neurons, which could add up to 1 quadrillion (1,000 trillion) connections (Brotherson, 2009).
Neurons have three basic parts: a cell body and two extensions called an axon and a dendrite. Nerve cells communicate via synapses, the space between neurons. Scientists estimate that in the brain’s communications network, one neuron may have as many as 7,000 synaptic connections with other neurons.
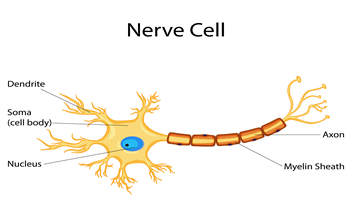
When a neuron receives signals from other neurons, it generates an electrical charge that travels down the length of its axon and releases neurotransmitter chemicals across the synapse. Like a key fitting into a lock, each neurotransmitter molecule then binds to specific receptor sites on a dendrite of a nearby neuron. This process triggers chemical or electrical signals that either stimulate or inhibit activity in the neuron receiving the signal.
Neurons have evolved to live a long time—more than 100 years in humans. As a result, neurons must constantly maintain and repair themselves and their synaptic connections. Adult brains may even generate new neurons from stem cells—a process called neurogenesis. Remodeling of synaptic connections and neurogenesis are important for learning, memory, and possibly brain repair (Gage, 2000; Conover & Notti, 2008).
Glial Cells
The term glia, derived from the Greek word meaning glue, reflects the nineteenth-century view that these cells had the function to hold the nervous system together (Virchow, 1856). In the brain, glial cells outnumber nerve cells 10 to 1. Glial cells provide physical and metabolic support to neurons, holding them in place, and helping them function as they should, including neuronal insulation, communication, and nutrient and waste transport.
Signals from glial cells are essential for brain development as they control the generation of neurons (Lim & Alvarez-Buylla, 1999), their survival (Gomes et al, 2001), and the migration of their somata and axons (Chotard & Salecker, 2004; Nadarajah & Parnavelas, 2002). Without glial cells, developing nerves often lose their way and struggle to form functioning synapses (Freeman, 2006).
Glial cells, in the central nervous system (CNS) comprise astrocytes, microglia, and oligodendrocytes. Importantly, each cell type performs critical functions that sustain the health and function of the surrounding neurons.
-Astrocytes
Astrocytes are the most abundant cells in the brain, outnumbering neurons by over fivefold (Sofroniew & Vinters, 2010). Astrocytes are star-shaped and anchor neurons to their blood supply which allows astrocytes to relay signals. Astrocytes play essential functions in blood brain barrier maintenance, neuronal survival, and in synapse formation, strength, and turnover, and they are critical for the regulation of cholesterol in the neuron. They also act as metabolic sensors in the brain responding to changes in the local environment, by removing excess ions, recycling neurotransmitters, and clearing synapses to prevent neurotoxicity (Wosik et al, 2007; Barres, 2008; Alvarez et al, 2011; Bell et al, 2012; Garcia-Caceres et al, 2016; Jäkel & Dimou, 2017).
-Microglia
Microglia are located throughout the brain and spinal cord and account for 10–15% of all cells found within the brain (Gilchrist 2021). Microglia are the resident macrophage cells, surrounding and removing dead cells, pathogens, and cellular debris, functioning as a sort of ‘clean up’ team. In this capacity, they also act as the first and main form of immune defense, responding to inflammatory stimuli and maintaining a stable environment (Akiyama et al, 1994). Microglia actively surveil and respond to the state of synapses, sensing dysfunction and helping repair or remove the cell (Graeber & Streit, 2010; Paolicelli et al, 2011).
Importantly, microglia can respond to inflammation. After the exposure of several stimuli, microglia release various neurotrophic factors, such as cytokines and reactive oxygen species (ROS) to promote neuronal cell survival. However, if the stimulation constantly occurs, microglia will be overactivated and steadily release the proinflammatory and cytotoxicity factors. This aberrant production of proinflammatory mediators can damage local cells, and further contributes to the neuroinflammatory process and is linked to neuronal degeneration (Streit et al, 2004; Akiyama et al, 2000). Together, this response regulates the course of the immune system’s response to inflammation, and whether brain homeostasis can be maintained (Sofroniew, et al, 2010; Nimmerjahn, et al, 2005).
-Oligodendrocytes.
The primary role of oligodendrocytes is to produce myelin, which ensheaths and insulates the axons of neurons. The myelin is essential to ensure rapid electrical conduction and neuron signaling, which underpins the massive computing power of the human brain (del Rio-Hortega, 1928; Nave & Werner, 2014). Myelination begins in the fourth month of human embryonic development and continues until the third or fourth decade of life (Brody et al, 1987; Lebel & Beaulieu, 2011). In general, the spinal cord and brain stem myelinate earlier, while other areas, such as the telencephalon, the entorhinal cortex, hippocampus and the amygdala myelinate later (Braak & Braak, 1996; Desai et al, 2009).
The adult central nervous system contains mature myelinating cells as well as precursor cells for oligodendrocytes which are capable of proliferating, migrating and effecting new myelination after demyelinating insults (Bauman & Pham-Dinh, 2001).
Myelin loss has been observed consistently in Alzheimer's Disease. Ideally, remyelination should be an ongoing process. However, the inflammatory milieu surrounding the demyelinated lesions compromises and limits the efficacy of the remyelination process (Franklin & Goldman, 2015). The disruption in myelin results in the demise of axons and neurodegeneration. The later-myelinated areas demonstrate significantly greater myelin loss compared with areas that myelinate earlier (Reisberg et al, 1999; Bartzokis, et al, 2003; Mitew et al 2010; Benitez, et al, 2014; Gao, Cheung et al, 2011). Myelin loss and the inability of the oligodendrocytes to repair myelin damage may be central features of Alzheimer's Disease (Matute, 2010; Matute et al, 2007; Pak et al, 2003).
Blood Brain Barrier (BBB)
The blood–brain barrier was discovered in the late 19th century, when the German physician Paul Ehrlich injected a dye into the bloodstream of a mouse. To his surprise, the dye infiltrated all tissues except the brain and spinal cord, showing that a barrier existed between the brain and the blood stream.
The blood–brain barrier is the interface between the vascular system and the brain, and functions as a barrier between the brain’s blood vessels and the cells and other components that make up brain tissue. Whereas the skull, meninges and cerebrospinal fluid protect against physical damage, the blood–brain barrier provides a defense against disease-causing pathogens and toxins that may be present in our blood.
The barrier consists of a highly selective, semipermeable border of endothelial cells in the capillaries, coated with transporter proteins, wedged extremely close to each other, forming tight junctions. The tight gap allows only ions, small molecules, fat-soluble molecules, and some gases to pass freely through the capillary wall and into brain tissue. Some larger molecules, such as glucose, can gain entry through transporter proteins, which act like special doors that open only for particular molecules.
The BBB is responsible to maintain the homeostasis of the brain by protecting against circulating toxins or pathogens that could cause brain infections, while at the same time allowing vital nutrients to reach the brain, hence transport from the bloodstream to the brain must be tightly regulated (Hawkins & Davis, 2005; Abbott et al, 2006; Abbott et al, 2010; Larsen et al, 2014; Wong, et al, 2013; Daneman & Prat, 2015; Rhea & Bank, 2019). The concentrations of water, ions, amino acids, hormones, and neurotransmitters in the blood undergo fluctuations, particularly after eating or exercise. If such fluctuations were allowed to occur in the brain it would lead to disruption of signals and uncontrolled neural activity. Only a small number of molecules such as alcohol and caffeine have been found to cross the blood brain barrier.
Dysfunction of the BBB can lead to ion dysregulation, altered signaling homeostasis, as well as the entry of immune cells and molecules into the CNS, processes that lead to neuron dysfunction and degeneration. BBB dysfunction is an important component of the pathology and progression of different neurological diseases (Zlokovic, 2008; Daneman, 2012).
While maintaining a stable environment for the brain, the selective permeability of the BBB remains a major roadblock to treatment of many central nervous system diseases, as many drugs are unable to pass the barrier.
Lipids in the Brain
Lipids are a class of molecules in the body that include hormones, fats, oils, and waxes. The nervous system of mammals has the highest lipid content (behind fatty tissue) and the most complex lipid profile of all bodily tissues. Brain lipids constitute 50% of the brain dry weight (O’Brien & Sampson, 1965; Hamilton et al, 2007; Luchtman et al, 2013; Bruce et al, 2017; Hannum & Obeid, 2018).
Lipids and lipid metabolism play an enormously important role in the structure and function of the cellular processes in brain tissue as well as brain health. The lipids act as signaling molecules, a source of energy, and contribute to the creation of synapses, neurons, impulse conduction and many other processes (Hussain, Schmitt et al, 2013; Cemenati, Mitro, et al, 2015).
All vital events responsible for the development and maintenance of the nervous system depend on the unique lipid contents found in the different membrane regions of neuronal cells. Any change in lipid metabolism results in changes in those lipids of the numerous membranes within cells, which is a common biomarker in many neuronal disorders (Adibhatla, Hatcher, 2008; Aureli, Grassi et al, 2015).
Lipids found in the brain are grouped as glycerophospholipids, cholesterol, and sphingolipids, and are present in almost equal ratios (Korade & Kenworthy, 2008; Zhang & Liu, 2015; Hussain, 2019)
-Glycerophospholipids
Glycerophospholipids are major constituents of cell membranes and are responsible for the membrane being a bilayer or having two layers. Marked changes in glycerophospholipid composition have been reported to occur in neurological disorders.
-Cholesterol
Cholesterol is a type of blood fat. Cholesterol and other lipids are carried in the blood attached to proteins, forming tiny spheres, or "parcels" known as lipoproteins. Cholesterol is essential for normal brain development and functioning (Zhang & Liu, 2015). Astrocytes are a major site of lipoprotein synthesis and assembly in the brain (Wang & Eckel, 2009).
Given its importance, highly sophisticated regulatory systems have evolved for the maintenance of cholesterol homeostasis in the body. The blood–brain barrier prevents entry of cholesterol-rich lipoproteins that are being carried in the bloodstream. Therefore, all cholesterol in the CNS is created in the brain (Turley et al 1996; Björkhem & Meaney, 2004). De novo synthesis is considered responsible for practically all cholesterol in the brain (Zhang & Liu, 2015).
(The fact that brain cholesterol metabolism is independent from that in the rest of the body warrants caution when causal correlations between high blood cholesterol and brain pathologies are suggested. With an intact blood–brain barrier, neurons and all other cells in the brain are not influenced by circulating cholesterol (Pfrieger, 2003; Brecht et al, 2004; Mahley et al 2006; Kiray et al, 2016).
The brain is the most cholesterol-rich organ, containing 25% of all cholesterol in the body (Allen et al, 2007). The majority of cholesterol present in the CNS is believed to reside in two different pools: one represented by the myelin sheaths and the other by the plasma membranes of astrocytes and neurons (Snipes & Suter, 1998). It has been estimated that up to 80% of the brain cholesterol is present in the myelin sheath.
Cholesterol helps guide developing nerve endings to their destinations on “lipid rafts”. If the brain is too low in cholesterol, its membranes, synapses, myelin and lipid rafts can't form or function properly, bringing all brain activity—including mood regulation, learning, and memory— to a screeching halt (Linetti et al, 2010; Liu et al, 2010).
Apolipoprotein E (ApoE) is a protein involved in the metabolism of fats and is synthesized in the brain which is the second largest producer of ApoE after the liver (Elshourbagy et al, 1985; Linton et al, 1991). The primary role for ApoE in the CNS is to shuttle cholesterol between cells (Vance & Hayashi, 2010; Hussain et al, 2019) and facilitate the efficient uptake of lipoproteins (Huang et al, 2011; Sterling et al, 2016).
A gene is a stretch of DNA that contains the instructions for making or regulating a specific protein. Inside individual cells some genes are active while others are not. Active genes are capable of producing proteins, called gene expression. Proteins form the internal machinery with cells and control the chemical reactions that allow brain cells to communicate with each other. Changes or differences in genes, called genetic variants, may increase or decrease a person's risk of developing a particular disease. When a genetic variant increases disease risk but does not directly cause a disease, it is called a genetic risk factor.
In the past decade, evidence of a possible link between neurodegeneration and cholesterol has accumulated. Some of the earliest observations of this link were the recognition of e4 isoform, or allele of ApoE as an important risk factor for late-onset Alzheimer disease (Burns & Duff, 2002; Mattson, 2004). An allele is one of two or more alternative forms of a gene that arise by mutation and are found at the same place on a chromosome. ApoE has two independently folded domains, and has three alleles known as e2, e3, and e4. This leads to six different genotypes: e2/2, e2/3, e2/4, e3/3, e3/4, and e4/4. These three Apo3 isoforms are profoundly different.
ApoE e4 is called a risk-factor gene because it increases a person's risk of developing the disease, particularly if you have two copies – e4/4. However, inheriting an ApoE e4 allele does not mean that a person will develop Alzheimer's. Some people with an ApoE e4 allele never get the disease, and others who develop Alzheimer's do not have any ApoE e4 alleles. ApoE is the only AD risk gene known to be involved in lipid metabolism (Agrawal et al, 2020).
About 25% of the people carry at least one copy of ApoE4, and 2-3% carry two copies. People inheriting one copy of e4 are three times more likely - but not guaranteed - to develop AD compared with people inheriting two e3 forms, whereas the ones having two copies of e4 are 8–12 times more at risk. In the general population only 10-15% will develop AD by age 85. Having the e2 allele partially protects against the disease (Oveisgharan et al, 2018; Agrawal et al, 2020).
Notably, the gene which often results in Alzheimer’s also results in a lower metabolic production of Vitamin K in the individual. In 2001, Allison observed that the circulating concentration of vitamin K1 is significantly lower and the incidence of Alzheimer’s significantly higher in ApoEr4 carriers (Allison, 2001). This inverse relationship results from the fact that vitamin K in plasma is bound to chylomicrons which carry ApoE. Clearance of chylomicrons from the circulation depends on ApoE’s binding to a hepatic receptor. People carrying one or two alleles for ApoE4 bind rapidly, and quickly depletes the blood levels of vitamin K available to circulate (Pizzorno, 2018).
-Sphingolipids
Sphingolipids were discovered in brain extracts in the 1870s and were named after the mythological sphinx because of their enigmatic nature (Thudichum, 1884; Chun & Hartung, 2010). Sphingolipids are a group of complex lipids primarily located in the nerve cell membranes, where they make up approximately 25% of the lipids in the myelin sheath (Sundaram & Lev, 1990). Major sphingolipids in the central nervous system include ceramides, sphingomyelin, cerebrosides, sulfatides, and gangliosides (Denisova et al, 2005; Posse de Chaves & Sipione, 2010; Olsen & Færgeman, 2017).
Sphingolipids are very important for brain function, and in the development and survival of neurons. Although originally appreciated as components of cell membranes, sphingolipids are now known to participate in important cellular events such as cell signaling, proliferation, differentiation and survival, cell growth, and death, cell-cell interaction, and cell transformation (Hakomori, et al 1991; Bell et al, 1993; Herr et al, 1997; Ariga et al, 1998; Cutler & Mattson, 2001; Ohanian & Ohanian, 2001; Levade et al, 2002; Zeidan & Hannun, 2007; Bartke & Hannun, 2009; Milhas et al, 2010; Gandy et al, 2013; Adada et al 2014; Yabu et al, 2015; Trayssac et al, 2018).
Sphingolipids are stimulated by vitamin K dependent proteins, which modulates their synthesis and metabolism. (Ferland, 2012; Chouet et al, 2019; Simes, et al, 2020). The involvement of vitamin K with sphingolipids metabolism, has recently gained renewed attention due to the role of sphingolipid metabolism in the aging process and neurodegenerative disorders such as Alzheimer’s disease (Cutler et al, 2004; Piccini et al, 2010; De Chaves et al, 2010).
Bone
A classic principle of physiology is that no single organ can develop alone (Qin et al, 2016). While the brain is a complex and powerful organ that plays essential roles in coordinating the body, the brain is also influenced by the feedback effects from other organs. A famous example is the microbioo-gut-brain axis (Vuotto et al, 2020). In recent years, bone has proven to be an endocrine organ, and the brain can exert regulation on the brain by secreting various molecules, several of which are essential to brain homeostasis (Chamouni et al, 2015; Downey et al, 2017; Yuan et al, 2019). A recent review of bone-derived modulators of the brain discusses the ones most researched and that these modulators could serve as potential molecular targets for the treatment of neurological disorders (Chen et al, 2021).
VITAMIN K
Primarily, there are two types of vitamin K, phylloquinone (K1) and menaquinones (MKs). K1 is the plant form of vitamin K, and it is primarily found in green, leafy vegetables. The next best sources are certain vegetable oils (e.g., soybean, rapeseed, and olive oils), with some in fruits, cereals and meats (Bolton-Smith et al, 2000; Thane et al, 2002; Popa et al, 2021).
Menaquinones or MKs account for only about 10–25% of the vitamin K content of Western diets (Schurgers et al, 1999; Nimptsch et al, 2008). Almost all the MKs are synthesized by bacteria. The highest food sources of long-chain MKs are animal livers and foods prepared with a bacterial fermentation stage such as cheeses (mainly MK-8 and MK-9) and sauerkraut, and natto (MK-7). Natto is a Japanese delicacy made by fermenting cooked soybeans, and it has a high content of MK-7 in a highly, bioavailable form (Schurgers & Vermeer, 2000). Although vegetables are 80-90% of the K1 intake, only 5–10% are absorbed, whereas MKs from dairy products are almost completely absorbed.
MK-4 is an atypical MK in that it is not commonly synthesized by bacteria but can be synthesized in vivo or locally. MK-4, is the only menaquinone that is obtained through conversion from VK1 and synthesized locally by brain tissues. Vitamin K participates in the brain as MK4 (Burt et al, 1977; Collins & Jones, 1981; Shearer & Newman, 2008; Nakagawa et al, 2010; Shearer, 2022).
Vitamin K is classically known for its role in the carboxylation and the biological activation of vitamin K-dependent proteins (VKDPs) necessary for the coagulation system. At least 19 proteins in the body have been identified as vitamin K dependent, meaning they need vitamin K to be activated or carboxylated. These carboxylated Gla proteins are found in many cell and tissue types throughout the body, including the brain. Carboxylation involves a transformation of the cell structure, with Glu residues transforming into Gla, which changes what the protein can do (Shearer & Newman, 2014). When carboxylated or activated, these proteins play roles in processes as diverse as bone and cardiovascular mineralization, vascular hemostasis, energy metabolism, immune response, brain metabolism, and in cellular growth, survival, and signaling. It is this very diversity of Gla proteins that makes vitamin K a true omni-vitamin (Popescu, 2018; Booth, 2009; Gundberg et al, 2012; Cancela et al, 2012; Schurgers et al, 2013; Ferland, 2012; Bellido-Martin et al, 2008; Laurance et al, 2012).
Vitamin K differs from other fat-soluble vitamins in that there are antagonists that block the synthesis of fully carboxylated Gla proteins. Coumarin or warfarin, well known as a clinical anticoagulant, also sold as a rat poison, interferes with vitamin K, blocking its protective action in the body, and the brain. A link to brain function was first reported in cases of central nervous system abnormalities in infants exposed in utero to vitamin K antagonists (Hall et al, 1980), which had blocked vitamin K during development (McCann et al, 2019).
Vitamin K has many complex roles in the brain through its activation of the K-dependent proteins present in the brain, and its role in sphingolipid synthesis, a major constituent of the myelin sheath and neuronal membranes [Ferland, 2012; Ferland, 2012; Chouet et al, 2015].
Vitamin K is a fat-soluble vitamin and is transported in plasma by lipoproteins. This means it is stored in the liver and fatty tissues. But unlike the other fat-soluble vitamins, the body stores very little vitamin K. This makes regular dietary intake very important (Lamon-Fava et al,1997).
Vitamin K Distribution
Vitamin K1, (phylloquinone) and MK4 (menaquinone) are differentially distributed in the body. Vitamin K1 is predominant in the liver and heart, while MK4 is predominant in the testes and brain. In an animal study, MK4 was found to represent more than 98% of the total vitamin K in the brain, irrespective of age (Carrie et al, 2004; Carrie et al, 2011; Shearer et al, 2012). Similar patterns of the tissue-specific distribution of vitamin K are observed in humans (Thijssen & Drittij-Reijnders, 1996; Benzakour & Kanthou, 2000; Tsaioun, 1999; Jolly et al, 1977].
MK4 is found in most tissues, though it occurs predominantly in the brain, identifying it as critically necessary for brain function (Thijssen, 1994; Huber et al, 1999). Within the brain, MK4 is found in all brain regions, although concentrations differ according to regions. Specifically, MK4 was found in highly myelinated areas, with the highest concentrations in the midbrain and pons medulla, and the lowest concentrations in the cerebellum, olfactory bulb, thalamus, hippocampus, and striatum (Carrie et al, 2004). A recent study by the Rush Memory and Aging Project analyzed post-mortem human brain samples. They found that MK4 was detected in more than 95% of brain samples from four brain regions (Fu et al, 2019). A later study found that higher MK4 concentrations in the brain were associated with a. 17%-20% lower odds of Braak stage with lower Alzheimer’s disease pathology score and fewer neurofibrillary tangles (Booth et al, 2021). (The Braak stage is a semiquantitative measure of neurofibrillary tangles).
Concentrations of MK4 in the brain are also affected by sex, age, and diet. In an animal study, MK-4 levels in the cortex and cerebellum were higher in female than in male rats despite similar diets, and concentrations decreased between 12 and 24 months of age (Huber et al, 1999). When rats were given low (80 ug per kg of body weight), adequate (500 ug per kg of body weight) or high (2000 ug per kg of body weight) amounts of K1 for 5 months, MK4 tissue concentrations from the high K1 diet were on average 8 and 3 times higher than those for the low and adequate K1 diets. Brain MK4 concentrations increased with vitamin K intake (Carrie et al, 2004).
Notable also is that MK4 is so essential that the blood brain barrier is not a barrier to it. The presence of MK4 in the brain, has been the object of study for fifty years, and currently it is believed that the human brain tissue obtains K1 from circulation and converts it to MK4 by the enzyme UBIAD1, with the synthesis taking place locally in the brain tissues (Nakagawa et al, 2010; Thijssen et al, 1996; Davidson et al, 1998; Thijssen et al, 2006; Okano et al, 2008; Hirota et al, 2013; Shearer & Newman, 2014).
A recent study of 48 participants from the Georgia Centenarian Study looked at vitamin K forms and their distribution in the frontal cortex (FC) and the temporal cortex (TC). The participants were a cohort of older adults whose cognitive status ranged from intact cognition to severe dementia, who participated in testing and who donated brain tissue upon death. They found that MK4 was the most predominant vitamin in all brain tissues, in both demented and non-demented subjects, accounting for ≥89.2% and ≥89.7% of total VK vitamers in FC and TC, respectively. They also found that circulating K1 concentrations were positively related to a wide range of cognitive tests among non-demented older adults. Serum/plasma K1 concentrations did not reflect brain MK-4 or total K concentrations (Tanprasertsuk et al, 2019).
Vitamin K Dependent Proteins - VKDP
Vitamin K participates in brain function through the VKDPs, Gas 5 and Protein S, which have been the most studied for their role in the brain (Stitt et al, 1995; Varnum et al, 1995; Nagata et al, 1996). Though, osteocalcin also has a role (Oury et al, 2013; Moser et al, 2018).
Tyrosine kinases are a family of enzymes which mediate cell signals. Receptor tyrosine kinases (RTKs) are the receptors on the surface of cells. RTKs play an important role in mediating cell-to-cell communication. When signaling molecules bind to RTKs, they cause neighboring RTKs to associate with each other, and interact with their neighbors in a tissue. TAM receptors are one subfamily of receptor tyrosine kinases (Burstyn-Cohen & Quan, 2021). TAM refers to Tyro3, Axl and Mertk, cell surface receptors that activate diverse downstream signaling pathways.
Research has established that Gas6 and ProS are both ligands of TAM receptors in the brain. Ligands are small molecules that transmit signals in between or within cells. Ligands exert their effects by binding to receptors. The ligand is like the baton, and the receptor is like the next runner in line. After binding to the ligand, the receptor can then send additional signals to other parts of the cell. The relationship between vitamin K dependent proteins and TAM receptors is critical for brain functioning.
Both TAM receptors (TAMRs) and their ligands, Gas6 and ProS, are widely distributed in the nervous system, and influence a wide range of cellular events (Lai & Lemke, 1991; Burstyn-Cohen et al, 2021). TAM receptor signaling modulates such events as neurogenesis and neuronal migration, cell proliferation, synapse flexibility, microglial activation, phagocytosis, myelination, and peripheral nerve repair. In turn, these events play complex roles in tissue repair, inflammation and cell survival, and migration (Prieto, 2000; Sainaghi et al, 2005; Hafizi & Dahlback, 2006; Rothlin et al, 2007; Cahoy et al, 2008; Chung et al, 2013; Han et al, 2013; Butovsky et al, 2014; Pierce & Keating, 2014; Lemke, 2013; Carrera-Silva 2013; Ji, Meng, et al, 2014; van der Meer et al, 2014; May, Garnett et al, 2015; Rothlin et al, 2015; Burstyn-Cohen, 2017; Fourgeaud et al, 2016; Healy et al, 2016; Shafit-Zagardo et al, 2018; Wium et al, 2018; Zhang & Qi, 2018).
The dysregulation of TAM signaling has been implicated in pathological processes leading to neuroinflammation, myelination abnormalities, neurodegeneration and injury to neurons. TAM deficiency impairs the neurogenesis of adult hippocampal cells, an important component of Alzheimer’s Disease (Gely-Pernot et al, 2012). Mice that were genetically deleted for all three TAM receptors had systemic chronic inflammation and autoimmunity, which led to brain damage, BBB breakdown, release of pro-inflammatory cytokines, protein aggregate formations and neuronal death (Li, et al, 2013). TAM receptors are linked to Alzheimer’s Disease and they may be a potential target for treatment (Tondo et al, 2019).
Gas 6
Gas6 is the product of Growth Arrest-Specific Gene 6, hence the name Gas6. It was discovered in 1993, and is a protein that depends on vitamin K availability to be carboxylated and activated. Gas6 appears to be the predominant vitamin K-dependent protein present in the brain (Prieto et al, 1999).
Gas6 is secreted by neurons and endothelial cells, produced in the cerebral cortex, hippocampus, cerebellum, midbrain and thalamus, and expressed by microglia, astrocytes and neural stem cells. This extensive expression in the CNS, points to its importance (Stitt et al, 1995; Prieto et al 1999; Gely-Pernot et al, 2012; Shankar et al, 2003; Binder et al, 2009; Pierce et al, 2014).
Gas6 expression decreases as a function of age, with the most dramatic change taking place in the frontal cortex. In a rat study, levels of Gas6 in the frontal cortex of 24-month-old rats were 84% lower than in 6-month-old rats, whereas in the striatum and hippocampus, the age-associated decrease was 55% lower (Tsaioun et al, 2000).
Gas6 is the main ligand for the TAM receptors, particularly the Axl receptor and a tantalizing assortment of roles for the Gas6–Axl system have been revealed. This is often referred to as the Gas6-Axl signaling axis, with Gas6 being the ligand (Lu et al, 1999; Ohashi et al, 1995; Stitt et al, 1995; Nagata et al, 1996; Mark et al, 1996; Hafizi & Dahlback, 2006). This ligand function depends on the presence of vitamin K (Varnum et al, 1995; Hall et al, 2002). Vitamin K carboxylation is the key to the interaction between Gas6 and the TAM receptors (Zuo et al, 2014).
Since its discovery, Gas6 has been associated with a wide range of cellular processes and has been shown to rescue cortical neurons from amyloid β-induced apoptosis, a hallmark of AD (Varnum et al, 1995; van der Meer et al, 2014; Ferland, 2012; Ferland, 2020).
-Gas6 - Cell Growth and Proliferation
Cell proliferation is the process of generating an increased number of cells through cell division.
Many studies have demonstrated the ability of Gas6 to promote either cell survival (Melaragno et al, 2004; van Ginkel et al, 2004) and/or proliferation (Stenhoff et al, 2004; Sainaghi et al, 2005). Working with mice where Gas6 was ‘knocked out’, (genetically removed), they found that the presence of Gas6 stimulated both DNA synthesis, and the proliferation of cells. Additional growth factor-like properties of Gas6 have also been reported, including stimulation of cell migration (Fridell et al,1998) and cell–cell adhesion via Axl (McCloskey et al,1997).
Schwann cells are named after German physiologist Theodore Schwann. There are two types of Schwann cells, myelinating and non-myelinating (Batheja & Field, 2006). Myelinating Schwann cells wrap around the axons of motor and sensory neurons to form the myelin sheath, which provides electrical insulation. A well-developed Schwann cell is shaped like a rolled-up sheet of paper, with layers of myelin between each coil.
Schwann cells are, are one of the principal components of the peripheral nervous system. They participate in the conduction of nerve impulses along axons, nerve development and regeneration, support for neurons, production of the nerve extracellular matrix, and modulation of neuromuscular synaptic activity. Gas6 has also been shown to induce the growth and proliferation of human Schwann cells (Li et al,1996).
Gas 6 – Mitosis
Cells are the building blocks of all living organisms. Cell growth refers to the increase in cell size. Cell division generates an increased number of cells through self-replication. Cell division is essential to life because it provides new cells for growth and for replacement of worn-out cells. Both cell division and growth are tightly connected and regulated. Cell division cannot generally be achieved without proper cell growth.
As part of cell division, mitosis is the division of the cell nucleus resulting in two genetically identical daughter cells, which allows DNA to be passed on. DNA contains genetic information of everything that is happening in one’s body, from the coding of an amino acid, to the color of one’s eyes. If our cells couldn't divide and create new cells, our bodies could never produce new skin cells to heal road rash or grow a fingernail back. Most importantly, genetic information would not be passed on. Research has shown that Gas6 has a mitogenic role, meaning that it induces a cell to begin cell division, or enhances the rate of division (Goruppi et al, 1996; Li et al, 1996; Ferland, 2012). This is accomplished through the Gas6/Axl pathway.
-Gas6 - Immunity
Tissue homeostasis and renewal requires both the birth of new cells and the death of old ones. In almost all settings, “out with the old” complements “in with the new.” Cells that are aberrant, aged, or infected must not only be killed but their corpses must also be efficiently cleared from tissues. The brain has developed a series of systems to recognize, dispose of, and recycle dead cells (Nagata et al, 2011).
This system features microglia, which are damage sensors for the central nervous system, and which function as phagocytes responsible for the routine clearance of dead brain cells. Phagocytes can engulf and absorb bacteria and other small cells and particles. Microglia, the ‘clean up team’ of the brain and spinal cord, are mobilized in response to nearly any CNS perturbation (Ransohoff & Cardona, 2010; Ginhoux et al, 2010).
Gas6 links with TAM receptors, which are specialized for the regular removal of apoptotic cells. Gas6 helps drive macrophages to recognize apoptotic cells and remove them (Scott et al 2001; Lemke et al, 2003; Hall et al, 2005; Wu et al, 2005; Lew et al, 2014). In the presence of Gas6, the uptake of dead cells by macrophages is enhanced (Ishimoto et al, 2000). When Gas6 is inhibited the removal of dead cells was inhibited (Sather et al, 2007).
Two recent studies using knock-out mice for Mer and Axl, demonstrated a reduced recruitment of microglial cells to sites of injury, and a reduced movement of microglial cells, which also affected the phagocytic activity, as the phagocytes lost their focus and directionality. This in turn led to an impairment in the clearance of dead cells. This highlights the important relationship between Gas6 and TAM receptors (Fourgeaud et al, 2016; Tang & Wu, 2015).
-Gas 6 and Anti-Apoptotic
Aoptosis is a form of programmed cell death. Apoptosis occurs normally during development and aging and is a mechanism to maintain cell populations in tissues and a homeostatic environment. Anti-apoptotic is when cells are helped to survive.
Gas6 has also been found to have anti-apoptotic effects on several CNS cell types such as cortical neurons, Schwann cells, oligodendrocytes, and in particular, hippocampal rat neurons (Allen et al, 1999; Allen et al, 2002; Funakoshi et al, 2002; Yagami et al, 2002; Pierce et al, 2008; Li et al, 1996; Nakagawa et al, 2002; Shankar et al, 2003, Shankar et al, 2006). In the nervous system, Gas6 has been shown to prevent cell death through activation of the Akt pathway (Li et al, 2019; Zuo et al, 2014).
Interestingly, cell culture studies have shown that Gas-6 modulates the survival of oligodendrocytes which produce myelin, preventing apoptosis or cell death (Binder & Kilpatrick, 2009). And notably, Gas6 protects the cortical neurons of mice from apoptosis induced by β amyloid protein, a defining feature of Alzheimer’s Disease (Yagami et al, 2003; Yagami et al, 2002).
-Gas6 and Remyelination
Myelination is vitally important to healthy central nervous system functioning. It allows more rapid transmission of neural information along neural fibers and is particularly critical in a cerebral nervous system that is dependent on several long axon connections between hemispheres, lobes, and cortical and subcortical structures. Remyelination is the regeneration of myelin sheaths following demyelination.
Research has established a role for Gas6 during remyelination. Mice genetically altered to be without Gas6, exhibited compromised oligodendrocyte survival, Others found that the deletion of Gas-6/Axl signaling led to prolonged neuroinflammation with axonal damage and consistent demyelination, accompanied by a reduction in overall myelination (Shankar et al, 2006; Binder et al, 2008). The administration of Gas6 to the corpus callosum enhanced the myelin repair in the same mouse model (Tsiperson et al 2010). In a later study, when Gas6 was added to a culture of oligodendrocyte cells, it resulted in an increased number of myelin segments in a dose dependent manner (Binder et al, 2011).
Research to better understand how the Gas6-Axl signaling axis affects axons during recovery from demyelination and inflammation, found Double Knock Out mice had significantly more axonal damage, fewer myelinated axons and significantly less myelin relative to regular mice. (Ray & Dubois, 2017). Collectively, results gathered have clearly established Gas6 as an important regulator of the myelination process.
-Gas 6 and Inflammation
Inflammation refers to the process whereby the brain's innate immune system is triggered by an injury, infection, exposure to a toxin, neurodegenerative disease, or aging. Neuroinflammation is common to neurodegenerative diseases and is often a cause of neuronal damage and cell death (Frischer et al, 2009; Guzman-Martinez et al, 2019; Kwon & Koh, 2020).
Microglia are the major immune cell in the CNS and work with astrocytes to maintain a stable environment, and to combat any challenge. Gas6 is highly expressed in microglia and present in astrocytes (Sofroniew & Vinters, 2010; Nimmerjahn et al, 2005; Binder et al, 2008; Shafit-Zagardo et al, 2018; Zhang et al, 2015).
The TAM (Tyro3, Axl, Mer) subfamily of receptor tyrosine kinases are key regulators of inflammation in the nervous system. The TAMs inhibit cytokine production and induce suppression of cytokine signaling, an element of inflammation. They also promote the survival of other CNS cells (Binder et al, 2008; Van der Meer et al, 2014; Hafizi et al, 2006). The vitamin K dependent protein Gas6,can alter signaling pathways associated with inflammation to resolve the inflammatory response within cells, reducing the possibility of chronic inflammation. Generally, Gas6 can bind and activate the Tyro3 and Axl receptors on astrocytes, and Mer and Axl receptors on microglia (Shafit-Zagardo et al, 2018). It appears that Gas6, through TAM receptor activation, is vital for reducing the inflammatory response (Olson & Miller, 2004; Sharif et al, 2006; Rothlin et al, 2007; Grommes et al, 2008; Sofroniew & Vinteres 2010; Alciato et al, 2010; Deng et al, 2012; Giangola et al, 2013; Rothlin et al, 2015) Kim et al, 2016; Peng et al, 2019; Gilchrist et al, 2020; Goudarzi et al, 2020; Gilchrist, 2021).
Alzheimer’s disease is associated with inflammation, and a recent study showed the upregulation of Gas6 and Protein S in the frontal cortex of AD patients, helped eliminate dead neurons and reduced inflammation. Gas6, acting through Axl and Mer, respectively, was involved in the phagocytosis of apoptotic neurons, something which can help prevent further degeneration (Herrera-Rivero et al, 2019].
-Gas6 and cognitive scores
Research has linked the presence of Gas6 with improved cognitive scores in patients with Alzheimer’s Disease. Elevated Gas6 levels were found in the cerebral spinal fluid of AD patients compared to controls, and the Gas6 levels were correlated with improved cognitive scores, suggesting Gas6 could play a defensive role in AD progression. Moreover, patients with higher Gas6 levels at diagnosis, displayed less cognitive deterioration over a two-year follow-up (Sainaghi et al, 2016).
-Gas6 and Amyloid β proteins
Amyloid β proteins lead to neuronal death both by promoting apoptosis and by direct toxicity, using a variety of mechanisms including the disruption of calcium homeostasis, oxidative stress, and mitochondrial dysfunction (Hadipour et al, 2020).
Very important research shows that Gas6 can rescue cortical neurons from apoptosis or death from amyloid β proteins, a key pathology in Alzheimers. Gas-6 was shown to decrease cell death induced by ß-amyloid and to reduce neurotoxicity (Ueda et al, 1997a; Ueda et al, 1997b), and the fragmentation of DNA was reduced (Yagami et al, 2002; Yagami et al, 2003; Mattson, 2004). It appeared that amyloid β proteins cause death via an influx of calcium into neurons, and the presence of Gas6 significantly inhibited the Aβ-induced Ca2+ influx, and the neurotoxicity (Hadipour et al, 2020). Evidence showed that Gas6 regulates activation of microglia, and helps stimulates the phagocytic response, which reduced the inflammatory response (Grommes et al, 2008).
TAM signaling is understudied in the field of AD, however, a role for Tyro3 has been indicated. It has been shown that activation of Tyro3 by Gas6 protects cortical neurons in vitro from the apoptosis induced by Aβ. It also reduced the production of Aβ, as well as reducing the formation of amyloid beta plaques, and plaque formation patterns. When mice lacked Tyro3, they presented with a higher number of amyloid beta plaques (Zheng et al 2012).
New data from Huang and colleagues found the TAM system to be essential in helping microglial recognize and engulf amyloid plaques. The microglia employed TAM receptors, Axl and Mer, to seek out and then engulf Aβ plaques. Genetic ablation of Axl and Mer resulted in microglia that were unable to detect, respond to, organize, or dispose of amyloid beta plaques. Targeting early events such as Aβ plaques may represent a promising strategy to slow or prevent disease initiation (Huang, et al, 2021; Wilson & Andreasson, 2021). This was possible, due to vitamin K activating the proteins which link to the TAM system.
Protein S
Protein S is a vitamin K dependent protein. Protein S is named for Seattle, Washington, where it was originally discovered in 1977. ProS is synthesized by different cell types, and has been found in many extrahepatic tissues, including the brain, testes, spleen, heart, endothelium and bone (Suleiman et al, 2013). ProS is expressed in the brain to a lesser extent than Gas6.
ProS is expressed in neural stem cells, Schwann cells, neurons, astrocytes and microglia (Jamison et al, 1995; Stitt et al, 1995; Lemke, 2013; Phillips et al, 1993; He et al, 1995; Prieto, Weber et al, 1999; Zhong et al, 2010; Wang et al, 2011; Gely-Pernot et al, 2012; Butovsky et al, 2014; Zelentsova et al, 2016; Zelentsova-Levytskyi et al, 2017; Zelentsova et al, 2017; Butovsky et al, 2014).
As a ligand of TAM receptors, specifically Tyro3 and Mer, ProS has been involved in important cell functions, that help regulate various tissues (Tsou et al, 2014; Zhu et al, 2010). ProS is involved in cell proliferation and survival, phagocytosis of apoptotic cells, and activation of innate immunity (Stitt et al, 1995; Anderson et al, 2003; Gely-Pernot et al, 2012; Ginisty et al, 2015; Abboud-Jarrous et al, 2017; Dahlback, 2018; Guo et al., 2011; Prasad et al, 2006; Zhu et al, 2010).
-ProS and Neurogenesis
Neurogenesis is a process by which neurons are generated from neural stem cells (NSC) and their descendants. Neurogenesis is linked to proliferation (Zelentsova et al, 2017). Massive neurogenesis occurs prenatally, and the process continues to a lesser extent throughout adulthood (Ericksson et al, 1998; Emsley et al, 2005). Neurons are continuously produced in brains of adults due to self-renewing divisions (Gage, 2000; Seri et al, 2004; Ming & Song, 2005; Suh et al, 2007; Conover & Notti, 2008; Bonaguidi et al, 2011). The continuous generation of new neurons within the hippocampus was shown to contribute to higher functions such as learning and memory (Deng et al, 2010; Gould et al, 1999; Murai et al, 2014; Shors et al, 2001).
Over their lifetime neural stem cells are faced with many binary decisions: to stay quiescent or to proliferate, to differentiate or to self-renew, and finally once differentiated, whether to adopt glial or neural fates (Song et al, 2012). This proliferation generates more stem cells for future replenishment, as well as neurons and astrocytes necessary for proper brain function (Gage, 2000; Conover & Notti, 2008).
TAMs regulate the survival, proliferation, and differentiation of neural stem cells. In vitro studies showed that NSCs lacking TAMs have reduced growth and proliferation, delayed differentiation and increased apoptosis (Ji et al, 2014). Research has shown that ProS helps maintain stem cell quiescence and is also necessary for generating new neurons. Together Gas6 and ProS regulate SVZ cell growth. Depleting ProS leads to a dramatic decrease in stem cell proliferation (Zelentsova et al, 2016; Gely-Pernot et al, 2012).
-ProS and Neural Support
ProS was also identified as a neuroprotectant during ischemic brain injury. A study showed that ProS is upregulated in the rat sciatic nerve as early as 1-2 days following nerve injury, highlighting its importance in response to injury (Stitt et al, 1995). In lab studies, systemically administered ProS protected neurons during ischemic brain injury and hypoxia/reoxygenation injury. In an in vivo model of stroke, Pro S was found to significantly reduce brain infarction and edema volumes and to improve blood flow in the brains of treated mice and directly protected neurons from hypoxic injury, leading to a better neurological outcome (Liu et al, 2003; Guo et al, 2011). Zhu et al. showed a direct correlation between the inhibition of Tyro3/Akt signaling pathway and the hypoxic-induced death of hippocampal neurons, underlining a potential protective effect of protein S in cerebral infarct (Zhu et al, 2016).
A study with mice, with Protein S genetically knocked out, found that they developed necrosis of the nervous tissue, along with embryonic lethal clotting, lack of blood flow, and hemorrhages. It appears that Protein S is needed during brain development for protection of nervous and vascular tissue (Burstyn-Cohen et al, 2009; Saller et al, 2009).
Following up, they found Protein S, via the TAM receptor Tyro3, was critical for neuronal protective activity. ProS reduced acute brain injury and helped mitigate chronic neurodegenerative disorders. They felt their data supported the development of new ProS-based approaches for reducing acute brain injury and mitigating chronic neurodegenerative disorders (Zhong et al, 2010).
-ProS and Synaptogenesis
Synapses control the strength of the signals transmitted between neurons. Over time, a synapse may strengthen or weaken due to fluctuations in activity, commonly known as synaptic plasticity. Underlying these processes is a phenomenon known as long term potentiation (LTP) which strengthens synapses based on patterns of activity and contributes to learning and memory (Lynch, 2004). ProS has been found to play a role in synapse turnover and the continual remodeling of synaptic architecture in the brain (Chung et al, 2013).
-ProS and Inflammation
Protein S is a ligand for all TAM receptors, and TAMs mediate inflammatory responses and macrophages. Macrophages are a type of white blood cell that surrounds and kills microorganisms, removes dead cells and stimulates the action of other immune system cells. TAM receptors and ProS are expressed by immune cells, including macrophages and dendritic cells. This signaling axis dampens immune reactivity and contributes toward resolving inflammation through at least two distinct mechanisms: the molecular inhibition of pro-inflammatory cytokines and by the phagocytic clearance of apoptotic cells (Carrera Silva et al, 2013; Zagorska et al, 2014).
A recent study found that ProS was upregulated in macrophages as inflammation was being resolved. When ProS was ’knocked out” genetically, and unavailable, macrophages engulfed fewer dead cell remnants, which are key events in the termination of inflammation. The results indicated that PROS1 could provide a new therapeutic target for inflammatory and fibrotic disorders (Lumbroso et al, 2018).
Dendritic cells (DCs), named for their probing, 'tree-like' or dendritic shapes, are responsible for the initiation of adaptive immune responses and hence function as the 'sentinels' of the immune system. Dendritic cells drive T-cell activation as part of the immune system defense. The magnitude of dendritic activation must be precisely controlled, otherwise it can lead to pathological conditions characterized by overreactive immune responses, such as allergies, autoimmunity, and chronic inflammatory diseases (Coombes & Powrie, 2008; Lambrecht & Hammad, 2010). A recent study found that T-cells, once activated, produced ProS that signaled through TAMs to limit the magnitude of dendritic cell activation. Genetic ablation of ProS in mouse T-cells led to increased expression of stimulatory molecules and cytokines, and enhanced immune responses as well as increased colitis (Carrera-Silva et al, 2013).
A recent study sought to identify novel targets for the treatment of Alzheimer’s disease within genes related to amyloid precursor protein (APP) which are the beginning of AB plaques. Their data showed that ProS /Tyro3 could generate an enhanced immune suppressive response to inflammation triggered by beta amyloid in moderate stages of Alzheimer’s. They also concluded that Gas6 was part of another subsystem, acting through Axl and Mer that was associated with advanced stages of disease progression (Herrera-Rivero et al, 2019).
What we have presented is the relationship between the vitamin K dependent proteins Gas6 and ProS, as ligands for the TAM receptors’ vast signaling system that impacts core brain functions in all its extraordinary complexity. What the research indicates is that these proteins are essential to brain functioning, and since they need vitamin K to carboxylate and activate them, it boils down to the need to take vitamin K consistently and in adequate amounts to be sure that these system requirements are satisfied.
Aging is a normal physiological process, and the brain does change with age. Typically certain parts of the brain shrink, with a loss of volume and neurons, especially those important to learning and other complex mental activities. In certain brain regions, communication between nerve cells may not be as effective. Blood flow may decrease, and inflammation may increase. These changes in the brain can affect mental function, even in healthy older people (Elobeid et al, 2016; Peters, 2006).
Neurodegenerative Disease
Neurodegenerative disease refers to a diverse group of disorders, characterized by the progressive loss of neurons and function, in different systems of the brain or the spinal cord (Fu & Hardy, 2018). This degeneration affects many of your body's activities, such as balance, movement, talking, breathing, and heart function. Most neurodegenerative diseases, such as dementia, have no cure. Unlike primary cells from skin, the liver, or muscle, neuronal cells of the CNS do not regenerate after damage by disease, ischemia (deprivation of oxygen, glucose, or blood flow), or physical trauma.
Neurodegenerative diseases affect millions of people worldwide. The achievements of medicine, and public health efforts in reducing early- and midlife mortality from certain cancers, infectious diseases, and cardiovascular disorders mean that a larger number of individuals are aging and therefore susceptible to neurodegenerative disease by virtue of their survival (Ransohoff, 2016).
Dementia
Dementia is not a single disease; it’s an overall term - like heart disease - that covers a wide range of specific medical conditions. Disorders grouped under the general term “dementia” are caused by abnormal brain changes or damage to brain cells. These changes trigger a decline in thinking skills, also known as cognitive abilities, severe enough to impair daily life and independent function. They also affect memory, language, problem-solving, behavior, feelings and relationships.
Alzheimer's is the most common cause of dementia, and accounts for 60-80% of cases (Weller & Budson, 2018). The second most common cause is vascular dementia resulting from microscopic bleeding and blood vessel blockage in the brain. Mixed dementia describes those who experience the brain changes of multiple types of dementia simultaneously. Most neurodegenerative diseases are associated with various degrees of cognitive impairment, and the decline in cognitive function is often progressive, and profound (Rossor et al, 2016; Aarsland et al, 2017).
Recent animal and human studies showed that bioactive compounds could diminish the risk or delay the onset or progression of dementia (Rusu et al, 2020; Chauhan & Chauhan, 2020; Carillo et al, 2021). In this section, we will review the research showing how vitamin K can mitigate some of the degenerative processes and support brain health.
Alzheimer’s Disease
Alzheimer's disease (AD) was first described by Alois Alzheimer in 1907 and is the most common form of neurodegenerative disease (Bondi et al, 2017). The incidence of Alzheimer’s disease (AD) has risen considerably in recent years, and affects an estimated 6.2 million Americans, a number that is projected to more than double by 2050 (Alzheimer’s Disease Facts and Figures, www.alz.org; Patterson, 2018). The risk of AD dramatically increases in individuals beyond the age of 70.
Clinically AD can be classified into two subtypes. About 95% of AD patients are 65 years or older and are diagnosed with ‘sporadic’ AD, while 5% of AD patients carry rare genetic mutations associated with early onset, and ‘familial’ AD. There are many types of events that culminate in Alzheimer’s Dementia (AD), including head injuries, genetics, strokes, and other diseases such as diabetes or vascular disease, or obesity (Li et al, 2015). These events can all lead to neurodegeneration (Wang et al, 2017). Aging is one of the main risk factors for neurodegenerative diseases. Aging is associated with significant changes in the brain, which typically shrinks to some degree, but does not lose neurons in large numbers (Scheibel et al, 1975; Fjell et al, 2016; Melzer et al, 2021).
In Alzheimer’s disease, however, damage is widespread. At first, at a cellular level, Alzheimer’s disease typically destroys neurons and their connections in the hippocampus and the entorhinal cortex, parts of the brain involved with memory and the maintenance of higher cognitive functions (Serrano-Pozo et al, 2011). The entorhinal cortex is part of the medial temporal lobe memory system and is the gateway for information entering and leaving the hippocampal formation (Mattson et al, 2004; Ballard et al, 2011; Jembrek et al, 2015). That is why memory loss is often one of the earliest symptoms of Alzheimer's. Later, areas in the cerebral cortex responsible for language, reasoning, and social behavior are damaged. Over time, a person with Alzheimer’s gradually loses his or her ability to live and function independently, and ultimately, the disease is fatal.
-Amyloid Plaques
Neuropathologically, Alzheimer’s disease (AD) is characterized by the accumulation of misfolded, amyloid-β (Aβ) proteins that have accumulated in plaques between neurons. Amyloid is a naturally occurring protein formed from the breakdown of a larger protein, called amyloid precursor protein (APP). Abnormal cleavage of APP results in the formation of amyloid-β protein, densely packed with beta sheets, which form plaques. The plaques disrupt cell function. Of the amyloid proteins, Aβ42 is the most toxic (Selkoe, 1996; Gandy et al, 2005; Rushworth & Hooper, 2011; Larson & Lesne, 2012; Hefti et al, 2013; Nhan, et al, 2015; Viola et al, 2015; Wirths et al, 2004; Guillozet et al, 2003; Tanz, 2005; Liao et al, 2007; Chatterjee & Mudher, 2018; Livingston et al, 2017; Hussain et al, 2018; Garad & Edelmann, et al, 2021; Tiwari Atluri et al, 2019).
-Tau Tangles
Alzheimers is also characterized by the formation of tangles of the binding protein tau in the brain, which collect inside neurons. Healthy neurons, in part, are supported internally by structures called microtubules, which help guide nutrients and molecules from the cell body to the axon and dendrites. In healthy neurons, tau normally binds to and stabilizes microtubules, and participates in transport and neurotransmission. In Alzheimer’s disease, however, abnormal chemical changes cause tau to detach from microtubules and stick to other tau molecules, forming threads that eventually join to form tangles inside neurons. These tangles block the neuron’s transport system along the axon, which harms the synaptic communication between neurons. Abnormal tau accumulates in brain regions involved in memory (Wang et al, 1995; Iqbal et al, 2010; Querfurth & LaFerla, 2010; Sanabria-Castro et al, 2017).
As the level of beta-amyloid reaches a tipping point, there is a rapid spread of tau throughout the brain. This cascade results in damaged neurites and synapses, and generally culminates in neuronal death. It may also produce toxins and inflammatory cytokines that contribute to the neurodegenerative process and brain atrophy. Brain regions with plaques typically exhibit reduced numbers of synapses, and neurons associated with the plaques are often damaged (Mattson, et al, 2004; Guilozet et al, 2003; Reiss, Arain et al, 2018; Kinney et al, 2018).
-Proteins
The word "protein" comes from the Greek proteios, which means "first" or "foremost," reflecting their importance (Linderstrom-Lang, 1953). Proteins are large, complex molecules that play many critical roles in the body. They do most of the work in cells and are required for the structure, function, and regulation of the body’s tissues and organs. Proteins are made up of hundreds or thousands of smaller units called amino acids, which are attached to one another in long chains. There are 20 different types of amino acids that can be combined to make a protein.
Protein’s fold into a configuration, coded in their amino acid sequence. This configuration is typically an alpha helix, and it happens in microseconds (Pauling et al, 1951; Ashraf et al, 2014). When a protein becomes toxic, it can fold into a beta sheet. This is characteristic of the amyloid deposits found in Alzheimers. Unfortunately, the toxic configuration is often able to interact with other native copies of the same protein and catalyze them to transition into the same toxic state, which leads to cell impairment or death. Accumulation of misfolded proteins can cause disease (Dobson, 2002; Dobson, 1999; Reynaud, 2010).
Since the discovery of amyloid plaques in 1984, research on AD has almost exclusively focused on the pathological role of this small peptide. This led to a popular theory – the Amyloid Cascade, in which it was believed that the Amyloid B protein, was the causative agent for the ensuing Alzheimer’s pathology and the neurofibrillary tangles, cell loss, vascular damage, and dementia that followed (Hardy & Selkoe, 2002; Ricciarelli & Fedele, 2017). However, many trials targeting Aβ have produced many negative results, demonstrating that though this peptide might participate in the evolution of the disease, it may not be the pathogenic factor it was believed to be, (Gilman et al, 2005; Holmes et al, 2008; Winblad et al, 2012; Pasquier et al, 2016). Research is ongoing to better understand how, and at what stage of the disease, the various forms of beta-amyloid influence Alzheimer’s.
-Neuroinflammation
Neuroinflammation is both a major risk factor as well as a common feature of Alzheimer’s Disease (McGeer et al, 2016; Kinney et al, 2018). Neuroinflammation is a process regulated by microglia cells, whose job it is to recognize and eliminate any toxic component in the central nervous system, such as an amyloid plaque. The microglia cells secrete proinflammatory cytokines such as IL-1β, IL-6, and TNF-α (Mosher & Wyss-Coray, 2014). In normal conditions, once the toxic stimuli have been cleared, microglia shifts to the anti-inflammatory phenotype and secretes anti-inflammatory cytokines such as interleukins (IL-4, IL-10 and IL-18), brain-derived neurotrophic factor (BDNF) or nerve growth factor (NGF), whose role is to terminate the immune response and restore healthy functioning (Verhatsky et al, 2016).
However, under pathological conditions, microglia cells do not go back to their resting state, instead they adopt reactive states characterized by increased phagocytosis and increased expression of receptors, cytokines, chemokines, reactive oxygen species, and additional inflammation related molecules (Wolf et al, 2017). Chronic inflammation can become highly toxic, leading to neurodegeneration (McGeer et al, 1988; Rogers et al, 1996; Ransohoff, 2016).
In Alzheimers disease, the role of microglia was initially thought to be incidental, however, recent genome-wide association studies have established the concept that microglia play a central role in AD-related inflammation and are now identified as a potential therapeutic target.
-Mitochondria
Mitochondria produce ATP, which is a special molecule for energy called adenosine triphosphate (ATP). Mitochondria produce ATP through the electron transport chain to provide cells with the energy needed for survival, Thus, mitochondria are called the powerhouse of the cell. When cells are damaged, the electron transport chain complex is impaired, ATP synthesis is reduced, and the clearance of free radicals and ROS is obstructed, resulting in an imbalance of oxidative stress (Hou et al, 2012; Yun & Finkel, 2014; Raefsky & Mattson, 2017).
In 2004, Swerdlow and Khan (2004) proposed a ‘mitochondrial cascade hypothesis’, which resulted in a ‘vicious cycle’ being formed among mitochondrial dysfunction, Aβ deposition and autophagy dysfunction (Swerdlow & Khan, 2004). Many studies have confirmed that mitochondrial dysfunction is involved in Alzheimers disease. It begins with the accumulation of Aβ plaques, which leads to mitochondria damage and dysfunction. The damage to mitochondria results in an imbalance between the production of free radicals and the antioxidative defense mechanism, causing chronic oxidative stress (Redmann et al, 2016; Kerr, et al, 2017; Zhao et al, 2018; Lin et al, 2021)
Vitamin MK4 has a variety of potent neuroprotective functions. First, vitamin MK4 serves as electron carrier to transfer electrons in the mitochondrial transport chain which helps to maintain normal ATP production, and increase mitochondrial membrane potential (Vos et al, 2012; Simes et al, 2020). Second, vitamin MK4 is anti-inflammatory and inhibits antioxidant stress (Hadipour et al, 2018; Saputra et al, 2019; Yang et al, 2020). Third, vitamin MK4 may promote the clearance of damaged mitochondria by activating autophagy (Miyazawa et al, 2020), and is anti-apoptotic (Yu et al, 2016), Studies have found that when astrocytes are exposed to hypoxia, MK-7 pretreatment not only reduces neuroinflammation but also increases ATP production and inhibits ROS production (Nakajima et al, 1993; Yang et al, 2020].
However, studies of the relationship between mitochondria, vitamin K and AD are just beginning. A recent study looked at the protective effect of MK4 in vivo using a fly model. The results showed that vitamin MK4 improved locomotor abilities, prolonged lifespan and significantly decreased Aβ42 level, a toxic element of AD (Lin et al, 2021). The findings suggested that MK4 did this by activating autophagy, maintained autophagy flow, and rescued mitochondrial dysfunction.
Another study found that MK4 reduced neuronal death, inhibited toxic reactive oxygen species, helped retain the mitochondrial membrane potential, down regulated the expression of tau protein and alleviated mitochondrial damage, highlighting its role as a new antioxidative therapeutic (Shandilya et al, 2021). An in vitro study showed that vitamin MK4 reduces the Aβ-induced cytotoxicity and improves cell survival (Saputra et al, 2019). To study the signaling of MK4 and mitochondrial dysfunction, cells were treated with a degenerative nerve agent. The results showed that vitamin MK4 blocked the expansion of mitochondrial damage, promoted biogenesis and improved the dysfunction induced by oxidative stress. (Tang et al, 2022).
Vitamin K is one of the bioactive compounds that has gained importance in recent years for its broad health applications, beyond coagulation. The early research is promising for its role in mitochondrial dysfunction, suggesting it may be a valuable therapeutic approach for AD (Lin et al, 2021).
Vitamin K and Alzheimer’s Disease
The presence of vitamin K has been linked to many improvements in Alzheimer’s disease. In the brain it would be vitamin K as MK4 (Thijssen et al, 1996).
Vitamin K - Neuron Protection
Oxidative stress refers to elevated levels of reactive oxygen species (ROS) that are key signaling molecules and which play an important role in the progression of inflammatory disorders. ROS can be damaging to cells. Research found that vitamin K can prevent oxidative damage to oligodendrocyte precursor cells, which myelinate nerves and cortical neurons. The data showed that both Vitamin K1 and MK4 blocked cell death. The next phase looked at pretreatment outcomes and they found that pretreatment with MK4 was sufficient to provide complete protection against cell death from oxidative insult (Li et al, 2003).
I/R refers to Ischemia and reperfusion which is a pathological condition characterized by an initial restriction of blood supply and oxygen to an organ, followed by a resumption. Following ischemia, the damaged cells cannot function properly, due to compromised metabolism. A short duration of I/R can lead to neuronal death especially in the hippocampus, and can cause learning and memory deficits (Elmore, 2007; Fu et al, 2014; Bacigaluppi et al, 2010).
A recent study focused on the neuroprotective effects of MK-4 in hippocampal cells in rats following cerebral I/R insult. The findings showed MK4 administration had positive effects on almost all aspects of ischemic injury. Administration of high amounts of MK4 (400mg/kg) immediately and again 2 hours after I/R injury compensated for the damage, leading to improvements in anxiety-like behavior, short term and spatial learning, and memory impairment. Also, MK4 was able to diminish the increased total brain water content, cell death, inflammatory markers, and increased overall survival. Additionally, MK4 significantly elevated SOD levels in the hippocampus and reduced oxidative stress (Moghadam et al, 2020). (SOD refers to superoxide dismutase which prevent the formation of ROS.) This is a very important study, as it indicates that very high levels of MK4 can be extremely neuroprotective! As an example, a 160 pound person weighs approximately 72 kg. In this study, they would have received 28,800 milligrams of MK4. This is a very high dose but given that all MK4 is nontoxic at any level, this could be a very desirable treatment option!
Autophagy eliminates abnormal misfolded proteins by clearing the amyloid plaques (Zhao et al, 2010). This process is hindered in Alzheimer’s Disease, resulting in Tau and Aβ clumping, which leads to the formation of neurofibrillary tangles and senile plaques. A recent study explored the effects of MK4 on flies with Alzheimer disease. They showed that vitamin MK4 can activate autophagy and maintain autophagy flow, which contributed to the clearance of Aβ, and reduced the Aβ-induced neurotoxicity. The study also showed that MK4 improved locomotor abilities, prolonged lifespan and significantly decreased AB42 level, and increased the ATP level (Lin et al, 2021).
A lab study looked at the protective effect of vitamin MK4 on toxicity and oxidative damage induced by amyloid plaques. Cells were pretreated with MK4 (5-200 ug) for 4, 24 and 48 hours, and then exposed to amyloid plaques or hydrogen peroxide. They found that MK4 pretreatment significantly decreased the toxicity and reduced the levels of ROS, which could halt the progression of the disease (Hadipour et al, 2020). These are very important findings, as they indicate that ensuring the availability of MK4 is very protective against the damage caused by Alzheimer’s Disease.
A 2021 study reached similar conclusions in a lab study. Astroglia cells in rats were transfected to express Aβ, then treated with MK4. Upon an increase of MK4, the cells survived longer. This effect was reversible with the addition of warfarin, which interferes with vitamin K. The MK4 also reduced the numbers of ROS in a dose dependent manner, and as well as decreased the enzymes that moderate Aβ cell death in a dose-dependent manner. This protective effect is likely to inhibit the Aβ-mediated apoptosis (Huang, et al, 2021).
When considered together, these studies present a powerful argument that vitamin K functions as antioxidant, and preserves cells against the destruction by beta amyloid and the progression of Alzheimer’s disease.
Vitamin K- Inflammation
Neuroinflammation is one of the major elements of Alzheimer’s Disease (Morales et al, 2014; McManus & Heneka, 2017; Calsolaro & Edison, 2016; Kinney et al, 2018; Chen et al, 2018). The emergence of β-amyloid plaques initiates neuroinflammation that is mediated by microglia. Microglia elicit the expression of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α). On the one side, activation of microglia leads to reducing Aβ accumulation by increasing its phagocytosis, clearance and degradation, which prevents the formation of amyloid plaques in the brain, followed by a process of returning to a normal state. On the other side, if persistent and chronic, the release of pro-inflammatory cytokines is also persistent and chronic, and contributes to ongoing neuroinflammation and ultimately leads to further neurodegeneration (Streit et al, 2004; Jiang et al, 2012;Lyman et al, 2014; Liddelow et al, 2017; Rice et al, 2015; Wang et al, 2015; Perry et al, 2015; Spangenberg et al, 2016;
The anti-inflammatory activity of vitamin K has been demonstrated in several in-vitro and animal studies. In vitro, MK4 has been shown to limit the production of inflammatory marker Il-6 in cultured human fribroblasts (Reddi et al, 1995) and prostaglandins (Koshihara et al, 1993). In animal studies, MK4 has been observed to limit inflammation in models of encephalomyelitis (Moriya et al, 2005; Ferland, 2012).
The brain is especially vulnerable to oxidative stress. A wealth of evidence suggests that high levels of ROS are linked to cell death and a redox imbalance (Popa-Wager et al, 2013). Vitamin K as an antioxidant agent has been proposed. The reduced form of vitamin K (KH2) was shown to protect membranes by the direct uptake of (ROS), thus preventing a buildup of ROS (Mukai et la, 1993; Vervoort et al, 1997). An important player in this antioxidant activity is VKORC1, an enzyme in the vitamin K cycle, which is responsible for limiting the amount of intracellular ROS (Westhofen et al, 2011). Recently, another study evaluating the effect of vitamin K on human osteoblasts showed that vitamin K in conjunction with vitamin D3, prevented a redox imbalance by decreasing ROS levels. The highest effect was obtained with MK-7 (Ambrozewicz et al, 2019).
Work by Li in 2003 demonstrated that cell death caused by oxidative stress can be prevented by the presence of vitamins K1 and MK4 in cultured neurons and oligodendrocytes. Vitamin K1 and MK4 were shown to prevent cell death, by blocking ROS production and preventing ROS accumulation. This could represent an alternative anti-inflammatory mechanism. Both vitamin K1 and MK4 have been shown to have antioxidant properties (Li et al, 2003; Li, et al, 2009).
Another study looked at the protective effect of MK4 and free radicals, which are a form of ROS, and which can lead to oxidation, and damage cells. They found that MK4 decreased free radicals and ROS, proportional to the dose, i.e. increasing the treatment of MK4 of 2, 5, 10, and 20 μM, led to a 25%, 30%, 36% and 40% reduction in free radicals (Huang, et al, 2021). Based on these results, dietary intake of vitamin K may strengthen defense against antioxidative stress and strengthen the immune system.
One way to induce a rapid and robust inflammatory reaction in the CNS is to administer lipopolysaccharide (LPS), a protein which mimics systemic infection. Several in vitro and animal studies have shown that when LPS was used, vitamin K1 supplementation reduced the activation of inflammatory markers with a consequent decrease in the production of pro-inflammatory cytokines. Both vitamin K1 and K2 (MK-3, MK4, and MK7) were found to suppress the inflammation in the mouse model. MK4 was found to suppress IL-6 production with higher efficiency than vitamin K1 (Ohsaki et al, 2006; Ohsaki et al, 2010; Fujii et al, 2015). A later study exposed mouse microglia cells to LPS. Pretreatment with MK4 inhibited the production of the inflammatory cytokines and the upregulation of cytokines. MK4 had the optimum effect compared to K1 and MK7. The least effective was MK7 (Saputra et al, 2019). This study demonstrated the importance of MK4 being present and available to cells, as a way to prevent inflammation and the ensuing damage.
When astrocytes are deprived of blood flow or oxygen, they can generate inflammatory cytokines. In a recent study, astrocytes were pretreated with various levels of MK7, under normal and hypoxic (lack of oxygen) conditions. The MK7 reduced the inflammatory response by decreasing the proinflammatory cytokines. As well, Gas6 was markedly increased after pretreatment (Hadipour et al, 2020). Another study found that MK7 reversed the upregulation of proinflammatory cytokines caused by glial activation in rat astrocytes (Yang et al, 2020). And importantly, another lab study showed that vitamin MK4 protected against β-amyloid toxicity and improved cell survival. In that study, MK4 reduced the ROS formation and inhibited cell death, confirming the antioxidant effects of MK4. The protective effect was abolished if warfarin was added (Huang et al, 2021). Despite using different types of glial cells (microglia and astrocytes), and distinct forms of VMK4 (MK4 and MK7), both these studies both reached the conclusion that vitamin K as menaquinone suppresses the production of proinflammatory cytokines, and reduces neuroinflammation and neurodegeneration.
Other research looked at the response of Gas6 and ProS and their signaling pathways in rats treated with warfarin. Male rats received either K1 and MK4, or not. After one week, the rats were administered warfarin (and K1 to maintain coagulation function) for nine weeks. Those receiving warfarin showed decreased activity of the important proteins Gas6, PS, and decreased signaling activity, as well as decreased BDNF (brain derived neurotropic factor) expression, a molecule related to learning and memory. They found a marked impact of warfarin on the hippocampus. Further, the rats from the warfarin group presented with increased expression of inflammation markers. Supplementing their diet with MK4, normalized the activity of protein S and Gas 6, and downstream signaling activity and reduced the inflammation markers (Ferland et al, 2019).
Vitamin K- Sphingolipids
Sphingolipids are a group of lipids that make up the myelin sheath, and are mediators of cell function (interaction, proliferation, senescence, differentiation, and transformation) and vitamin K is involved in their synthesis (Carrie, et al, 2004; Ferland, 2012; de Chaves et al, 2010). Alterations in sphingolipid metabolism have been linked to the development of Alzheimer’s disease (Han, 2005; Denisova & Booth, 2005).
Work establishing a role for vitamin K in sphingolipid metabolism is largely based on the legacy of Meir Lev’s group. In a series of publications that spanned from 1958 through 1996, Meir Lev and colleagues (Lev, 1979; Lev et al, 1988) provided evidence that vitamin K modulates key enzymes of the sphingolipid pathway and hence their synthesis and metabolism. This evidence was first established in bacteria (Lev & Milford, 1972) and then in mice (Sundaram & Lev, 1988; Sundaram & Lev, 1990; Sundaram et al, 1996). Treating mice with warfarin decreased brain concentrations of sphingolipids, while providing an excess of vitamin K1 restored the balance. In rats, the stimulatory effect of vitamin K on the activity of sphingolipids enzymes was observed with either K1 or MK4 as a source of vitamin K (Sundaram et al, 1996). It was clear that vitamin K was essential for sphingolipid synthesis.
In the following years, work continued exploring the relationship between dietary vitamin K and sphingolipids in the brain. Isabelle CarrieÌ studied the distribution of vitamin K and sphingolipids in various brain regions in 6-mo-old female Sprague-Dawley rats who were fed a low, adequate, or high phylloquinone-containing diet since weaning. The concentration of MK-4 was found to positively correlate with the concentrations of sphingolipids, particularly with sulfatides and sphingomyelin and negatively correlated with gangliosides, indicated a moderating role for vitamin K. Both vitamin K1 and MK4 increased, with K1 intake. When phylloquinone was present in the diet in limited amounts, MK4 preferentially accumulated in highly myelinated regions. Furthermore, the strong negative correlation observed between MK4 and gangliosides suggested a potential modulatory role for vitamin K in the general sphingolipid pathway (Carrie et al, 2004).
Later the same team investigated learning abilities in distinct groups of 6-, 12-, and 20-month-old female rats that had been fed diets containing low (~80 μg/kg diet), adequate (~500 μg/kg diet), or high (~2000 μg/kg diet) levels of phylloquinone since weaning. Lifetime consumption of a low-vitamin K diet resulted in cognitive deficits in the 20-mo-old rats, with those in the low group having longer latencies than those in the high group; this was associated with higher concentrations of ceramides in the hippocampus and lower gangliosides in the pons medulla and midbrain. The low-vitamin K diet did not affect cognition at 6 and 12 months of age. The results also showed that dietary K1 altered brain sphingolipid concentrations in a significant manner, related to the different intakes of vitamin K. The rats that received the diet containing low amounts of phylloquinone throughout their lives (20 months) had significantly higher levels of ceramides, which is associated with cognitive impairment (Carrie et al, 2011). Ceramides, the major molecules of sphingolipid metabolism and lipid messengers, have been associated with AD progression and pathology via Aβ generation.
Studies of Alzheimer's disease show a significant decrease in brain sulfatide concentrations, which is another type of sphingolipid (Gottfries et al, 1996; Soderberg et al 1992; Cutler et al, 2004). A report on the very early stages of AD showed that sulfatides were decreased as much as 93% in gray matter and as much as 58% in white matter, whereas ceramides were increased threefold, suggesting that alterations in sphingolipid metabolism may be involved in the pathological process of AD (Han et al, 2002).
Research has shown that higher concentrations of MK4 in the hippocampus and cortex were shown to be correlated with desirably higher myelin sulfatides in adult and aged rats, but not in younger animals, suggesting that age is a factor (Crivello et al, 2010). In a study of mice that underwent demyelination an intake of 2 mg of K1 three times per week increased the production of brain sulfatides after remyelination. And the phylloquinone K1 treatment increased the levels of MK4 in the brain (Popescue et al, 2018). A new study using a mouse model showed that sulfatide losses in myelinating cells activated microglia and astrocytes, and increased the expression of genes associated with Alzheimer’s disease risk. This resulted in chronic neuroinflammation and mild cognitive impairment and was more pronounced in females than males (Qiu et al, 2021). These studies indicate that sulfatide deficiency is a contributor and driver of neuroinflammation and cognitive impairment in Alzheimers, and vitamin K raises the levels.
Vitamin K- and Cognition
There has been ongoing work investigating vitamin K and its impact on cognitive functioning, as a general representation of optimal brain functioning.
-Animal Studies
Animal studies pertaining to cognition are limited. One important study raised rats from birth on a diet that was low in vitamin K versus a diet that was adequate or high in K1, and then measured their learning pace or latency. They found that the older rats on a diet low in K1 needed more time to acquire spatial learning and needed more exposure to visual cues when compared to those on an adequate or high vitamin K diet. The low vitamin K diet had no impact on cognition in the younger animals, suggesting that vitamin K is very important to brain function in a vulnerable, aging state. Also, these cognitive alterations with age were associated with higher concentrations of ceramides in the hippocampus and lower gangliosides in the pons medulla and midbrain, similar to AD. The hippocampus is the brain region responsible for memory consolidation and an altered sphingolipid profile in that area bodes poorly for optimal brain function (Carrie et al, 2011).
A recent animal study showed that high doses of MK7 (30mg/kg) for 17 months had a positive impact on functional, behavioral, biochemical and histopathological scales. They displayed fewer anxiety or depressive behaviors, or memory deterioration. The oxidative load in the brain was reduced and there were significant improvements in the brain’s biochemical and histopathological inflammatory status. And age-related inflammation, or inflammaging, was evident (Elkattawy et al, 2022).
Keep in mind, the possible action of vitamin K in the hippocampus is also strengthened by studies on the VKD protein Gas6. Specifically, the receptor Tyro3, one of the Gas6 ligands, is highly expressed in the hippocampal region, and its activation by Gas6 results in molecular events known to underlie memory consolidation (Davis & Laroche, 2006; Horwood et al, 2006; Prieto et al, 2007). So, a low vitamin K diet would mean less activation of Gas6 and less signaling of the Tyro3 pathway in the hippocampus, and poorer memory function.
-Human Studies
Research has laid out the foundational role for Vitamin K in the nervous system. Evidence is also mounting for an expanded role for vitamin K and cognition. There is a developing body of research that has demonstrated, in a population of 65 years and older, a direct correlation between low vitamin K dietary intake and deteriorated cognitive and behavioral performances (McCann et al, 2019; Maresz, 2021).
An early study on the nutritional status of community-dwelling patients with early-stage Alzheimer’s disease found that average vitamin K intake was twofold less than that of control subjects (Shatenstein et al, 2007). The Nutrition-Memory study recruited early-stage Alzheimer dementia patients and age-matched them to a cognitively intact control group. They were assessed four to five time-points over a 12-to-18-month period and diet and supplements were assessed monthly. The results showed that nutrient intakes were higher in the control group, with significant differences in the intake of vitamin K (Carrie et al, 2004). It is conceivable that these Alzheimer dementia patients had poor diets before diagnosis, which could be a manifestation of the development of Alzheimer dementia (Rogers, 2001). As the findings showed fairly consistent nutrient intakes over the 1-year follow-up reported herein, it would appear that deterioration in food consumption occurs early in the disease, and once in place, remains a characteristic feature (Goodwin et al, 1983; Riviere et al, 2001; Priefer & Robbins, 1997; Rogers, 2001; Holm & Soderham, 2003; Noel & Reddy, 2005; Luchsinger, et al, 2004; Del Pariegi et al, 2006)
In an additional analysis of the Nutrition-Memory Study cohort, the same group found that 31 cases with mild to moderate Alzheimers disease had lower dietary vitamin K1 intake compared to a control group that were age and gender matched. The group with AD had an intake of about 37 units per day versus 70 units per day in the control group. The very minimum daily requirement is 90 units a day for women and 120 units a day for men, meaning the patients in the AD group were significantly below the requirement. It was almost a 55% difference in K1 intake. These findings suggest that a low vitamin K intake could be a risk factor for Alzheimer’s disease. Although patients could have underreported their food intakes because of their mild cognitive impairment, the data collection method limited this possibility (Presse et al, 2008).
Accordingly, an epidemiological study reported a significant association between higher serum phylloquinone concentration (vitamin K1) and better verbal episodic memory performance in older adults (Presse et al, 2013). In that study, they examined the associations between K1 concentration and verbal and non-verbal episodic memory, executive functions, and speed of processing in 320 healthy older adults selected from the Québec Longitudinal Study on Nutrition and Successful Aging (NuAge). At the time of the cognitive evaluation, participants were aged 70.0 to 85.5 years and had normal cognitive functioning. The study showed that serum phylloquinone was positively associated with performance in verbal episodic memory, while being unrelated to non-verbal episodic memory, executive functions, and speed of processing. Episodic memory refers to the memory of events within their spatio-temporal context. For example, remembering where one’s keys were last left relies on episodic memory. Episodic memory processes include encoding (when the information to memorize is first encountered) and memory consolidation (when the memory trace is stabilized for long-term storage). The findings advocate for a specific role of vitamin K in memory consolidation. This pattern indicates that the higher the vitamin K status, the more the participants benefited from the successive learning trials.
A later study looked at the relationship between vitamin K intake, cognition and behavior among 192 older adults, age 65 years and over, recruited in the cross-sectional CLIP (Cognition and LIPophilic vitamins) study at the University Hospital of Angers, France, from February to April 2014. The participants were separated into two groups according to the quantity of dietary phylloquinone intake (i.e., lowest third below 207 µg/day versus the other two thirds combined). The main finding was that, irrespective of all measured potential confounders, increased dietary phylloquinone intake was associated with better cognition and behavior among geriatric patients (Chouet et al, 2015).
Furthermore, another study showed that increased vitamin K intake was associated with less frequent and severe subjective memory complaints in older adults. One hundred sixty older adults taking no vitamin K antagonist were included. The daily dietary vitamin K intake and subjective memory complaint were assessed at the same time. They found that increased dietary vitamin K intake was associated with fewer and less severe subjective memory complaints in older adults taking no vitamin K antagonists. Those with serious subjective memory complaints had a lower mean dietary vitamin K intake than those with normal memory (Soutief-Veillon et al, 2016).
The ELDERMET cohort is a well-defined study group of Irish adults, aged 64+ years, ranging from cognitively intact to severely cognitively impaired. This multifaceted project commenced in 2007 to examine how intestinal bacteria influence, and are influenced by, diet, lifestyle and health. In the current study, relationships between cognitive function, vitamin K status and inflammation were investigated. Subjects were divided into four groups based on their cognitive score. They found that both dietary intake and blood levels of K1 were significant and independent predictors of cognitive function. As well, there were significant differences in dietary phylloquinone intake and the inflammatory marker IL-6 between cognitive function groups, with low intake being associated with higher levels of inflammation. Even after controlling for inflammation, serum phylloquinone remained a significant and independent predictor of cognitive function (Keily et al, 2020).
NHANES is the National Health and Nutrition Examination Survey, a program conducted by the Centers for Disease Control and Prevention (CDC) to collect data on the health and nutritional status of adults and children in the United States. An analysis of their data showed that low dietary intake of vitamin K was significantly associated with a low cognitive score in the elderly. The lower the intake, the greater the risk of low cognition (Wang et al, 2022).
Likewise, results from a mature population, 65 years and older, revealed a direct correlation between low vitamin K dietary intake and low serum vitamin K concentration, as well as declined cognitive performances. In 2020, a total of 800 people (mean age = 75.9) residing in the Itabashi ward of metropolitan Tokyo were invited to a comprehensive geriatric health examination and a blood test. They demonstrated the association of vitamin K insufficiency as evaluated by serum uncarboxylated osteocalcin, (another VKDP) with cognitive impairment in the older adult population. The highest range of ucOC was associated with impaired orientation, calculation, and language. They concluded that vitamin K insufficiency could be associated with selected categories of cognitive function (Azuma et al, 2021; Alisi et al, 2019).
A recent study aimed to characterize the forms of vitamin K and their distribution in the frontal cortex and temporal cortex in a cohort of older adults enrolled in the Georgia Centenarian Study. 320 participants, aged 70-85 years, with and without cognitive impairment, were enrolled. Participants were approached for the opportunity to participate in the brain donation, and 66 participants agreed to donate brain tissues upon death. After enrollment, a battery of cognitive tests was administered at baseline and every 6 months until mortality. Blood and matched brain samples from the frontal and temporal lobes were collected. They found that MK4 was the most predominant vitamer in all subjects accounting for ≥89.2% and ≥89.7% of total VK vitamers in both the FC (Frontal cortex) and TC (Temporal cortex). Only MK4 and K1 were detected and quantified with our analysis. Whereas MK4 was detected in all samples, K1 was not detected in 39% of FC samples and 55% of TC samples. Circulating K1 concentrations were not associated with cerebral MK4 or total VK concentrations, regardless of dementia status. Circulating K1 concentrations were positively related to a wide range of cognitive tests among nondemented older adults. In a group of nondemented centenarians, only circulating K1 levels were significantly linked with a wide range of cognitive performance, specifically better verbal episodic memory performance. Even though MK4 was the predominant isomer in both the frontal and temporal cortex, cerebral MK4 levels were not associated with cognitive measures (Tanprasertsuk et al, 2020).
An animal study just published looked at the effects of MK7 on cognition. Three-month-old rats were given MK7 for 17 months, and their cognitive performance was compared to a control group who did not receive the MK7. The results showed that rats who received the MK7 performed better on cognitive tests. Analysis of the data showed that pathways associated with inflammation and antioxidant activity, and an amino acid that prevents cognitive decline, were positively affected by MK7. The amount of MK7 was 30mg/kg, which is a high dose. MK7 improved functional performance, reduced social anxiety, depressive-like behavior, and enhanced memory performance while preserving hippocampal and cerebral cortex expression. Biochemically, MK7 administration restored oxidative-anti-oxidative homeostasis in the brain and modulated inflammatory signaling. Concomitantly, histopathological examination revealed consistent hippocampal and cerebral cortex improvement. Thus, it can be inferred that high intakes of MK7 can slow down age-related changes in the brain (Elkattawy et al, 2022).
Additional indirect evidence stems from two cohort studies in which the high consumption of green leafy vegetables, rich in vitamin K, was associated with slower rates of cognitive decline. In the Nurses Health Study, nurses were followed for decades. The data showed that women consuming the most green leafy vegetables also experienced slower cognitive decline than women consuming the least amount of food that contained vitamin K1 (Kang et al, 2005). Another study followed 960 participants of the Memory and Aging Project, ages 58–99 years, living in the community, who completed a food frequency questionnaire and had more than 2 cognitive assessments over a mean 4.7 years. The data showed that the consumption of green leafy vegetables was linearly associated with slower cognitive decline. The rate of decline among those who consumed 1–2 servings per day was the equivalent of being 11 years younger compared with those who rarely or never consumed green leafy vegetables (Morris et al, 2006; Morris et al, 2018).
Vitamin K Deficiency and Alzheimer’s Disease
Research has established a strong link between a diet that is deficient in vitamin K and Alzheimer’s Disease. This was first hypothesized by Allison, in 2001. Given that the dietary intake of vitamin MK4 and the MK4 concentration in the blood stream are decreased in the circulating blood of apolipoprotein E4 (ApoE4) carriers, she argued that supplementing with MK4 may have a beneficial effect in preventing or treating AD (Allison, 2001).
An early study on the nutritional status of community-dwelling patients with early-stage Alzheimer’s disease found that average vitamin K intake was twofold less than that of control subjects (Shatenstein et al, 2007). The Nutrition-Memory study recruited early-stage Alzheimer dementia patients and age-matched them to a cognitively intact control group. The results showed that nutrient intakes were higher in the control group, with significantly lower differences in the intake of vitamin K in the group with Alzheimer’s disease (Carrie, et al, 2004).
In an additional analysis of the Nutrition-Memory Study cohort, the same group found that 31 cases with mild to moderate Alzheimer’s Disease had lower dietary vitamin K1 intake compared to a control group that were age and gender matched. The group with AD had an intake of about 37 units per day versus 70 units per day in the control group. The major result of this study is that patients in the early stages of probable Alzheimer’s disease were found to have significantly lower vitamin K intakes than age- and sex matched healthy participants, suggesting that insufficient vitamin K intakes could be a risk factor (Presse et al, 2008).
The National Heart, Lung and Blood Institute Twin study revealed that carriers of ApoE had significantly smaller brain volumes. The presence of both cerebrovascular disease and ApoE4 was associated with significantly greater brain atrophy. The authors concluded that ApoE4 enhances the extent of brain degeneration by increasing the brain’s susceptibility to injury and/or impairing brain repair mechanisms (DeCarli et al, 1999). One mechanism may be the insufficiency of vitamin K, whose concentration is lowered by ApoE4 and whose actions improve neuron survival and repair after brain injury.
Warfarin as vitamin K Deficiency
Warfarin is a widely used anticoagulant drug that is prescribed to millions of patients worldwide. It has long been used as the main drug for the prevention and treatment of blood clots for patients with prosthetic heart valves, for A-fib, and for the prevention of myocardial infarction and stroke. Warfarin and its derivatives decrease blood coagulation by inhibiting the vitamin K cycle, rendering vitamin K inactive and causing a relative “vitamin K deficiency (Božina 2010; Alquwaizani et al. 2013). They are referred to as vitamin K antagonists (VKA).
The impact of warfarin was studied in an animal model, with respect to cognition, behavior, MK4 and sphingolipid status. Fourteen male rats were treated with 15 mg W/kg/d (in drinking water) and subcutaneous K1 (85 mg/kg), 3 times a week, for 10 weeks; 14 control rats were treated with normal water and injected with saline. At the end of the treatment period, rats were subjected to different behavioral tests, after which their brains were assessed for phylloquinone and MK4, and sphingolipids. Those who received warfarin treatment showed a dramatic decrease in MK4 concentration in all brain regions, altered sphingolipid level, especially in the frontal cortex and midbrain, and a loss of sphingolipid regional differences, notably for gangliosides. The warfarin-induced vitamin K deficiency was associated with cognitive impairment, hypoactivity and lower exploratory behavior in rats (Ferland et al, 2013).
This same research group sought to confirm the role of vitamin K as it relates to sphingolipids, cognition, and behavior outside the context of aging, so they created a study of acute vitamin K deficiency, versus a lifetime of deficiency. In this procedure, rats were maintained on a ratio of warfarin with some K1, so that coagulation was maintained while inducing VK deficiency. After 10 weeks of treatment, rats who were given the warfarin plus phylloquinone protocol (WVK) exhibited longer latencies in visual learning as well as lower locomotor activity and exploratory behavior, when compared to control rats. The WVK treatment resulted in a dramatic decrease in MK-4 level in all brain regions despite the presence of high local concentrations of phylloquinone, which suggests a specific inhibition of the MK4 pathway in the presence of warfarin. Additionally, WVK treatment affected sphingolipid concentrations in key brain regions, notably those of the ganglioside family. In conclusion, this study provides further evidence that targeted depletion of MK4 in brain is associated with cognitive impairment, lower locomotor activity, and exploratory behavior, and with an alteration of sphingolipids in key brain regions, Results also suggest that, in vivo, phylloquinone in brain is not bio-transformed into MK4 in the presence of warfarin. In the control group, MK4 represented about 85% of the total vitamin K while in the experimental group which received warfarin, MK4 was only 20% of the total vitamin K (Tamadon-Nejad et al, 2018).
Given VKAs are commonly used anticoagulants in older persons, we investigated the relationship between VKA therapy and cognitive performances over 10 years in participants of the Three-City study, who lived in Bordeaux, Dijon, or Montepellier, France. They had to be 65 years or more when enrolling. VKA therapy, and platelet aggregation inhibitors (PAI) and cognitive performance were assessed every two years, over a period of 10 years, which included 7,133 nondemented community dwellers age 65 or older. The results showed that VKA treatment was significantly associated with lower performances on tests assessing visual memory and verbal fluency. Treatment with VKAs was not associated with global cognitive functioning nor with rate of subsequent decline in scores on all three cognitive tests. In cross-sectional analyses at baseline, older adults treated with VKAs, but not those treated with PAIs, had significantly, although clinically modest, lower performance in visual working memory and verbal fluency compared to individuals receiving neither antithrombotic treatment (Ferland et al, 2016).
Other research has shown that geriatric patients who take vitamin K antagonists (VKAs) as anticoagulant medications show cognitive impairment. A study at the Angers University Hospital, France, with the WARHOL study, explored whether VKAs were associated with cognitive decline among geriatric patients. They found that the risk of cognitive impairment was 15% higher with VKAs, specifically fluindione, an anticoagulant widely used in France (Annweiler et al, 2015; Annweiler et al, 2015a). WARHOL is an acronym for (Who is At Risk of Hypovitaminosis in Older individuaLs). A 24 month follow-up study found that the use of VKAs were associated with executive dysfunction in older adults. Executive functions refer to a heterogeneous set of high-level processes that regulate other abilities and behaviors. From a neuroanatomical point of view, executive functions are based on the integrity of frontal-subcortical circuits (Godefroy et al, 2008). This study showed that more frequent and severe executive decline was associated with the use of VKAs among geriatric outpatients (Brangier et al, 2018).
Another study from this group looked at community-dwelling individuals from the GAIT study (Gait and Alzheimer’s Tracking). They found that the use of VKA by older adults was associated with a smaller brain volume, specifically, lower volumes of gray matter were found, including in the temporal cortex and hippocampus. In a cross-sectional study conducted among geriatric patients, taking VKA was significantly associated with a higher risk of having cognitive impairment (Annweiler et al, 2015; Ferland et al, 2016). A recent study of patients using VKA for more than three months showed more intracranial calcifications than those who did not use VKAs (Annweiler et al, 2020).
Another study with subjects (≥75 years) on VKAs demonstrated that there is an association between VK1 concentration and cognition. At the University of Rome, 85 patients on anticoagulant therapy, between the ages of 75 and 92, were enrolled. Cognitive measures were positively correlated with levels of vitamin K1 concentration. The cognitive capacities did not correlate with the long-term use of anticoagulants (Alisi et al, 2020).
Taken together, this research suggests the importance of adequate (i.e., high enough) vitamin K levels for optimal cognition in older adults.
Vitamin K and Brain Injury
AD is the largest cause of dementia, causing 60–70% of cases worldwide (Tarawneh & Holtzman, 2012). Although the pathophysiology of AD is not completely understood, increased prevalence of AD is seen among patients who experienced traumatic brain injury, suggesting AD is linked to CNS injuries and inflammation (Fleminger et al, 2003; Li, Li, Li, et al, 2017).
Traumatic brain injury, stroke, hypoxic or ischemic injury and nerve crush can all result in inflammation, a milieu of cell death and neurological damage. TAM signaling molecules play a role in the recovery process. Gas6 and PROS1 both support neuronal survival under stress conditions, indicating the need for sufficient vitamin K to be available (Funakoshi et al, 2002; Zhong et al, 2010).
Neurofilament light chain (NfL) constitutes the backbone of nerve fibers. It is a protein that is released into the cerebrospinal fluid and blood when neurons are injured. Lower levels are good. The higher the level, the higher the risk of dementia (Cronje et al, 2023). NfL has been proposed as a biomarker of neurodegenerative diseases (Bacioglu et al, 2016). Dietary vitamin K intake has been found to be negatively associated with serum NfL levels, meaning high K intake was associated with lower NfL levels and reduced risk of brain disease (Luo & Lin, 2024).
Alzheimer’s Disease and Vascular Disease
People with dementia seldom have only Alzheimer’s-related changes in their brains. Any number of vascular issues—problems that affect blood vessels, such as atherosclerosis (hardening of the arteries), and mini-strokes—may also be at play. Vascular problems may lead to reduced blood flow and oxygen to the brain, as well as a breakdown of the blood-brain barrier, which usually protects the brain from harmful agents while allowing in glucose and other necessary factors. In a person with Alzheimer’s, a faulty blood-brain barrier prevents glucose from reaching the brain and prevents the clearing away of toxic beta-amyloid and tau proteins. This results in inflammation, which adds to vascular problems in the brain. Because Alzheimer’s is both a cause and consequence of vascular problems in the brain, researchers are seeking interventions to disrupt this complicated and destructive cycle.
Cardiovascular disease (CVD) has been shown to play an important role in the etiology of AD, and epidemiological studies have established that vascular disease increases AD risk (Newman, et al, 2005; Qui, Winblad et al, 2003; Toledo et al, 2012; Kapasi & Schneider, 2016; Kelleher et al, 2013; De laTorre, 2010; De Bruijn & Ikram, 2014). Cerebral atherosclerosis, small vessel disease, cerebral amyloid angiopathy, and blood-brain barrier dysfunction have all been reported in AD (Grinberg & Thal, 2010).
Arterial stiffness, arteriolosclerosis and endothelial dysfunction are characteristic of cerebrovascular disease, and have been known to contribute to neurovascular disintegration, brain atrophy, and the accumulation of cerebral Aβ (Kalaria et al, 2012; Zlokovic, 2011).
Additionally, aortic stiffness has been associated with an increased risk of dementia (Cui et al, 2018). And another study found a relationship between arterial stiffness, blood pressure, and the progression of beta amyloid in nondemented elderly adults (Hughes et al, 2014).
In a population-based cohort study of 844 patients, the amount of atherosclerotic calcification was directly proportional to cognitive impairment and inversely related to brain tissue volume (Bos et al, 2012). Furthermore, a greater volume of calcification, as measured by a computed tomography (CT) scan, corresponded to reduced integrity of white matter. Three years later, a large population study that followed 2364 participants for five years found that atherosclerotic calcification was associated with an increased risk of developing dementia (Bos et al, 2015). A cohort study with 1732 participants reported a similar finding and established an association between generalized atherosclerosis and mild cognitive impairment (Weimar et al, 2015).
One study demonstrated that carotid artery calcification was associated with a higher risk of dementia, and a recent cohort study that followed 4988 middle-aged Dutch ImaLife participants over a 10-year period showed that coronary artery calcium severity was associated with cognitive decline (Di Daniele et al 2019; Xia et al, 2021).
Interventions improving vascular function also have attenuated AD pathology [Kalaria et al, 2012). One study of 2907 elderly individuals demonstrated that the APOE gene serves as a genetic link between AD and vascular disease (Lin et al, 2019).
Autopsies of individuals with dementia revealed that 80% of cases had AD, 7–10% had vascular dementia, and 3–5% had mixed dementia, with vascular lesions found in 20–40% of the subjects who had AD (Jellinger, 2002). Another study involving autopsies found an association between vascular risk factors, cerebrovascular disease, and AD among 5715 subjects (Toledo et al, 2013). In addition, data have suggested that vascular and neurodegenerative processes can coexist in AD (Attems, 2014).
A recent animal study, found that vitamin K can oppose the detrimental effects of vascular calcification on cognition by limiting calcification, restoring resting cerebral blood flow and increasing Gas 6 and Protein S carboxylation in the brain. They gave the rats high doses of vitamin K, either .1, 1, or 10 mg/kg of vitamin K1 or 10, 50 or 100 mg/kg of MK4 for 4 weeks. In the groups that received lower amounts of vitamin K, their memory retention was altered by calcification. And regardless of diet, MK4 was the main type of vitamin K found in the brain. Expression of carboxylated Gas6 and PS increased with dietary MK4 but not dietary K1. Carotid calcium content decreased as a function of dietary K1 and MK4 in a dose dependent manner. The resting cerebral blood flow decreased in several brain regions of mice fed the low K1 0.1 mg/kg diet, including visual and somatosensory cortex, thalamus, hippocampus, dentate gyrus and corpus callosum (Ferland et al, 2020).
We have extensively reviewed the research on vitamin K and vitamin MK4 on vascular health in general. To read that, please go to https://www.k-vitamins.com/index.php?page=research-view-all&id=19
Alzheimer’s Disease and Diabetes
In the Diabetes page of this website (https://www.k-vitamins.com/index.php?page=research-view-all&id=150 ) we have extensively reviewed the research on how vitamin K helps with diabetes. Now we will discuss the research linking diabetes to Alzheimer’s disease.
Type II diabetes (T2DM) is a risk factor for, and a comorbidity in AD. A growing body of epidemiological studies show that Type 2 diabetes increases the risk of AD by at least 2-fold (Craft, 2007; Sims-Robinson et al, 2010; Cheng et al, 2011; Biessels, et al, 2014; Barbagallo & Dominguez, 2014). Longitudinal studies showed that patients with adult-onset diabetes exhibited a significantly higher risk of developing AD than age-matched subjects without T2DM (Leibson et la, 1997; Huang et al, 2014). According to the Mayo Clinic Alzheimer Disease Patient Registry, 80% of AD patients show impairment in glucose tolerance or have diabetes (Janson et al, 2004).
The exact connection between Alzheimer’s disease (AD) and type 2 diabetes is still in debate. However, poorly controlled blood sugar may increase the risk of developing Alzheimer’s. Many recent studies provide evidence that deficits in insulin signaling, arising due to insulin resistance, occurs in AD (Talbot et al, 2012; Mullins et al, 2017). Studies of the brains of “early stage” AD patients have demonstrated reduced glucose uptake. This relationship is so strong that some have called Alzheimer’s “diabetes of the brain” or “type 3 diabetes (T3D)” (De la Monte & Wands, 2008; Nguyen et al, 2020). Type 3 diabetes has been defined as a metabolic syndrome linked to progressive brain insulin resistance with consequent impairment of insulin signaling processes, accumulation of neurotoxins, neuronal stress, and resulting in neurodegeneration (Nguyen et al, 2020; Caberlotto et al, 2019).
Due to the large size of insulin peptide, it was thought that insulin is unable to cross the brain blood barrier (BBB), such that the brain was regarded as an insulin-independent organ (Banks, 2004). However, several studies have revealed that insulin receptors are abundantly distributed throughout the brain, proposing some mechanisms to explain the existence of insulin in the brain (Gray et al, 2004). Currently, insulin has been shown to be transported across the BBB through a carrier-mediated, active process (Medhi et al, 2013; Kang & Rivest, 2012).
Insulin is a crucial factor controlling blood glucose levels and it facilitates cellular glucose uptake in peripheral tissues by activating the PI3K-Akt pathway (Kim & Feldman, 2012). In the brain, insulin does not have a major role in glucose metabolism. However, insulin and the PI3K-Akt signaling pathway play a significant role in neuronal health as well as synapse formation and maintenance (Chui et al, 2008; Lee et al, 2011). While not fully understood the PI3K-Akt signaling pathway plays a crucial role in Type 2 diabetes and has also been observed in Alzheimer’s disease (Gabbouj et a, 2019). While most of the studies have reported no association, two studies have found a significant correlation between peripheral insulin resistance and brain Aβ levels as measured by PET scans (Willette et al, 2015; Ekblad et al, 2018).
Furthermore, alterations in the insulin signaling pathway as well as decreased levels of insulin and insulin receptors (IR) have been observed in the AD brain. Similarly, to T2D, other abnormalities such as metabolic stress and inflammation are also characteristic in AD (Steen et al, 2005; Moloney et al, 2010; Talbot et al, 2012). However, the relationship between T2D and the main neuropathological finding in AD, cerebral Aβ accumulation, remains unclear.
Insulin resistance leads to loss of synapses, impaired autophagy and increased neuronal apoptosis. These alterations might trigger a cascade of events leading to abnormal Aβ and tau accumulation culminating in Alzheimer's disease pathology.
As the brain is likely to encounter defective insulin signaling with increasing age (Cole & Frautschy, 2007), researchers have begun to study mechanistic links between these diseases. The impaired insulin-PI3K-Akt signaling observed in the AD brain has led to clinical trials studying whether the enhancement of this pathway using intranasal insulin (IN) treatment is beneficial (Steen et al, 2005).
Vitamin MK4 and the Gut Microbiome
There is a wealth of literature on the gut microbiome and its relationship to disease (Collins et al, 2012; Westfall, et al 2017; Kim et al, 2020; Srikantha et al, 2019; Holsinger et al, 2020; Sanada et al, 2020). The gut is the primary organ responsible for the digestion and absorption of nutrients. There is a saying that “it’s not what you eat, but what you absorb”, highlighting the importance of the gut.
An emerging area of research is the role of the gut microbiome in brain health (Collins et al, 2012; Magnusson et al, 2015). The brain-gut-microbiome axis is a two-way communication system that allows gut microbes to communicate with the brain and the brain with the intestine. Changes on either side of this axis may cause changes to the other. The signaling between these two systems and the microbiome are complex, involving metabolic, immune, neuronal, and endocrine signaling pathways, and are just being explored (Harach et al, 2017).
An emerging hypothesis is that the gut microbiota may influence AD neuroinflammation. It has been proposed that a microbial dysbiosis or imbalance at the gut level may impair gut permeability, thus inducing a systemic activation of the immune system, leading to AD. (Lai et al, 2021).
Trends in the research involve looking at the role of fecal transplants (Hazan et al, 2020; Xu & Wang, 2016; Holsinger et al, 2020; Kim, et al, 2020; Tran, et al, 2019), the role of antibiotics in altering the gut microbiome (Minter, et al, 2016), the administration of probiotics (Akbari et al, 2016; Leblhuber et al, 2017; Athari et al, 2018; Sanborn et al, 2018; Chen et al, 2017), inflammation in the gut (Cattaneo et al, 2017; and the loss of microbiome diversity with age (Westfall et al, 2017; Garcia-Pena et al, 2017) as contributors to disease.
There is emerging literature on vitamin K and the gut. A 2019 study revealed that antibiotic-triggered disruption of the mice microbiome decreased not only bone strength and bone density, but also the number of microbial genes involved in MK synthesis (Guss et al, 2019). As a result, there was also a decrease in the amount of MK4 in the cecum, liver, and kidney. These findings are supported by a study that showed that antibiotic treatment in mice fed a diet low in vitamin K eradicated the gut bacteria that produce menaquinone, leading to vitamin K deficiency and gastric hemorrhage. RNA sequencing confirmed a decrease in the menA and menD genes, which are involved in MK4 biosynthesis (Quinn et al, 2020). In another study, decreased levels of MK4 in SIBO (small intestinal bacterial overgrowth) were associated with increased arterial calcification and subclinical atherosclerosis (Ponziani et al, 2017).
An important study was carried out in Ireland by McCann and Jeffery. The Eldermet Cohort was established in 2007 to investigate the interaction between the gut microbiota, diet, and health in a cohort of older Irish individuals. Subjects ranged in age from 64–93 years, and were categorized based on where they resided, long term care, community, rehab, or day hospital. Cognitive abilities were assessed as well as levels of vitamin K1, inflammatory markers, and microbiota diversity. Researchers measured the fecal menaquinone levels and sorted them into groups based on the genetic potential of their microbiome to biosynthesize menaquinones. They found that variation and diversity in the MK biosynthesis concentration was associated with significant differences in cognitive levels (McCann et al, 2019), with more variation being associated with higher cognitive scores. Those with the lowest variation of genes that synthesized and produced MKs, were typified by a number of negative health parameters (McCann & Jeffery, 2019).
The gut microbiome has a vital role in physiology, pathology, aging and neurodegenerative diseases, and it is an exciting frontier! The possible link between Alzheimer’s disease and the gut has become the focus of intense interest in biomedical research, however the exact relationship between neurodegenerative diseases and gut microbiota remains unclear. At this point, is important to underline that most of the studies on human microbiota are observational and report just an association between a specific disease of interest and a microbial alteration. Currently, the challenge is to move to a more mechanistic view. In this context, different mechanisms are being investigated to clarify how the gut microbiota may influence the nervous system, including short-chain fatty acid secretion, blood–brain barrier permeability modifications, vagus nerve stimulation, or neurotransmitter modulation (Erny et al, 2015; Braniste et al, 2014; Strandwitz, 2018).
Summary
Alzheimer’s Disease is a significant concern for an aging population. Large pharmacological trials have failed to find an effective medication to treat or prevent AD, and there has been a shift towards research on nonpharmacological interventions. As of May 2022, the number of nonpharmacological studies (132) was almost double the number of pharmacological ones (73) (NIA Funded Trials; https://www.nia.nih.gov/research/ongoing-AD-trials).
The research presented here is clear. Vitamin K activates proteins, Gas6 and Protein S, which are distributed throughout the brain. These proteins connect to and activate vast signaling networks throughout the brain, the TAMs. Research shows that these networks affect cell division, cell growth, cell preservation, myelination, synapse development, support the brain’s immune system, and are part of the response to injury and damage, and fight inflammation. Additionally, these proteins are linked to improved cognitive scores and specifically rescue cells from the damage created by amyloid B plaques and tau disruptions. Very exciting are the studies showing that very high doses of vitamin MK4 can greatly ameliorate damage from strokes and infarcts, with no side effects. Vitamin MK4 has the potential to slow the progression of AD and contribute to its prevention (Popescu & German, 2021).
Animal studies show that a diet low in vitamin K is associated with learning delays and brain profiles that resemble Alzheimer’s disease. Longitudinal and epidemiological studies show that many folks with Alzheimer’s disease have a significantly low intake of vitamin K, and folks with a chronic vitamin K deficiency performed lower on memory tasks. The intake of vitamin K even predicted levels of cognitive function. In animal studies, warfarin, which interferes with vitamin K has been shown to decrease the amount of MK4 found in all brain regions, which was associated with cognitive impairments. In human studies, the use of warfarin (aka vitamin K deficiency) was associated with lower performance in visual working memory, verbal fluency, general cognitive impairment, a decline in executive function, more intracranial calcifications, and smaller brain volume. Given the roles of vitamin MK4 in the context of AD, and, given the recent shift in AD research toward nonpharmacological interventions, there has been calls for clinical studies involving vitamin MK4 (Popescue & German, 2021).
Numerous studies have shown that the presence of vitamin K supports a healthy brain and reduces the impact of disease. It can’t be stressed enough that the recommended daily allowance (RDA) for vitamin K is based on the minimum requirements for your coagulation system. The RDA does not reflect or account for any of the VKDP in tissues and organs throughout the body or in the brain that need vitamin K to be functional, and a diet based on the recommended current levels has actually been shown to decrease activation of VKDP (DiNicolantonio et al, 2015). Since dietary intake as well as the bioavailability of vitamin K from food are both low, supplementation of vitamin K should be considered for a number of chronic conditions, especially among elderly people (Schwalfenberg, 2017). Vitamin K is regarded as a promising agent in Alzheimer Disease therapy (Visioli et al, 2016.
As summarized in this paper, research has established a relationship between vitamin MK4 (MK4) and many elements of Alzheimer’s disease, including amyloid neurotoxicity, neuroinflammation, mitochondrial dysfunction, cognition, cardiovascular health, the microbiome, and AD comorbidities.
The unavoidable conclusion is that consistent and sufficient vitamin K intake benefits your brain functioning and can prevent or mitigate damage from injury, or a disease like dementia.
References
1880-1970
Virchow, R. 1856. Gesammelte Abhandlungen zyr Wissenschaftlischen Medizin. Frankfurt (A.M): Verlag von Meidinger Sohn & Comp.
Thudichum JLW. 1884. The chemical constitution of the brain. London (Eng): Baillère, Tindall, and Cox.
Del Río Hortega P. 1928. Tercera aportación al conocimiento morfológico e interpretación funcional de la oligodendroglía. Mem. Real Soc Esp Hist Nat. 1928;14:5–122.
Pauling L, Corey RB, Branson HR. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. PN AS. 1951 April;37(4):205-211.
Linderstrom-Lang KU. How is a protein made? Scientific American. 1953;189(3):100-107.
O’Brien JS, Sampson EL. Fatty acid and fatty aldehyde composition of the major brain lipids in normal human gray matter, white matter, and myelin. J Lipid Res. 1965 Oct;6(4):545-51.
Lev M. Vitamin K deficiency in Fusiformis nigrescens. I. Influence on whole cells and cell envelope characteristics. J Bacteriol. 1968;95:2317–24.
Lev M, Milford AF. Vitamin K stimulation of sphingolipid synthesis. Biochem Biophys Res Commun. 1971;45:358–62.
Lev M, Milford AF. Effect of vitamin K depletion and restoration on sphingolipid metabolism in Bacteroides melaninogenicus. J Lipid Res. 1972;13:364–70.
Scheibel M.E., Lindsay R.D., Tomiyasu U., Scheibel A.B. Progressive dendritic changes in aging human cortex. Exp. Neurol. 1975;47:392–403.
Jolly DW, Craig C, Nelson Jr, TE. Estrogen and prothrombin synthesis: effect of estrogen on absorption of vitamin K1. Am J Physiol. 1977 Jan;232(1):H12-7.
Burt VT, Bee E, Pennock JF. The formation of menaquinone-4 (vitamin K) and its oxide in some marine invertebrates. Biochem. J. 1977;162: 297-302.
Lev M. Sphingolipid biosynthesis and vitamin K metabolism in Bacteroides melaninogenicus. Am J Clin Nutr. 1979;32:179–86.
1980
Hall JG, Pauli RM, Wilson KM. Maternal and fetal sequelae of anticoagulation during pregnancy. Am J Med. 1980;68(1):122–40.
Of 418 reported pregnancies in which coumarin derivatives were used, one-sixth resulted in abnormal liveborn infants, one-sixth in abortion or stillbirth and, at most, two-thirds in apparently normal infants. In addition to the expected hemorrhagic complications, fetal effects of coumarin derivative administration include a specific embryopathy and central nervous system abnormalities.
Collins MD, Jones D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implications. Microbiol Rev. 1981;45:316-354.
Goodwin JS, Goodwin JM, Garry PJ. Association between nutritional status and cognitive functioning in a healthy elderly population. JAMA. 1983 Jun 3;249(21):2917-21.
We evaluated the association between nutritional status and cognitive functioning in 260 noninstitutionalized men and women older than 60 years who had no known physical illnesses and were receiving no medications. Nutritional status was evaluated by three-day food records and also by biochemical determination of blood levels of specific nutrients. Cognitive status was evaluated. Subjects with low blood levels of vitamins C or B12 scored worse on both tests. Subjects with low levels of riboflavin or folic acid scored worse on the categories test. These differences remained significant after controlling for age, gender, level of income, and amount of education. "Subclinical" malnutrition may play a small role in the depression of cognitive function detectable in some elderly individuals, or depressed cognitive function may result in reduced nutrient intake.
Brody BA, Kinney HC, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J Neuropathol Exp Neurol 1987;46:283–301.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291.
Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82:203–207.
Lev M, Sundaram S. 1988. Modulation of glycosphingolipid synthesis by vitamin K depletion in bacteria and brain. In: Suttie JW, editor. Current Advances in Vitamin K Research: Proceedings of the 17th Steenbock Symposium. Madison: University of Wisconsin-Madison.
Sundaram KS, Lev M. Warfarin administration reduces synthesis of sulfatides and other sphingolipids in mouse brain. J Lipid Res. 1988;29:1475–9.
1990
Sundaram KS, Lev M. Regulation of sulfotransferase activity by vitamin K in mouse brain. Arch Biochem Biophys. 1990;277:109–13.
Sundaram KS, Lev M. Vitamin K and phosphate mediated enhancement of brain sulfotransferase activity. Biochem Biophys Res Commun. 1990;169:927–32.
Lai C., Lemke G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. 1991;6:691–704.
Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, Boyles JK, Wardell MR, Young SG. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88:270–281.
Giusto, N. M., Roque, M. E. & Illincheta de Boschero, M. G. Effects of aging on the content, composition and synthesis of sphingomyelin in the central nervous system. Lipids. 1992;27:835–839.
Mukai K., Shingo I., Morimoto H. Stopped-flow kinetic study of Vitamin E regeneration reaction with biological hydroquinones (reduced forms of ubiquinone, vitamin K and tocopherolquinone) in solution. Biol. Chem. 1992;267:22277–22281.
Soderberg M, Edlund C, Alafuzoff I, Kristensson K, & Dallner G. Lipid composition in different regions of the brain in Alzheimer's disease/senile dementia of Alzheimer's type. J. Eurochem. 1992;59:1646–1653.
Bell RM, Hannun YA, Merrill AH, Jr. editors. 1993. Advances in Lipid Research: Sphingolipids and Their Metabolite.,, Vols. 25 and 26,. San Diego, California: Academic Press.
Koshihara Y, Hoshi K, Shiraki M. Vitamin K2 (menatetrenone) inhibits prostaglandin synthesis in cultured human osteoblast-like periosteal cells by inhibiting prostaglandin H synthase activity. Biochem Pharma. 1993;46(8):1355-62.
Nakajima M, Furukawa S, Hayashi K, Yamada A, Kawashima T, Hayashi Y. Age-dependent survival-promoting activity of vitamin K on cultured CNS neurons. Develop Brain Res. 1993;73:17-23.
Neurons from the central nervous system (CNS) of rat embryos die within several days when seeded at a low density of 104 cells/cm 2 and cultured in a serum-free defined medium. Using these culture systems, we searched for agents to promote the survival of these neurons. As a consequence, vitamin K1, was found to possess such kind of activity: more than 50% of the cortical neurons from 19-day-old rat embryos could survive for 4 days in the presence of vitamin K 1 whereas almost all neurons died in its absence. The survival-promoting effect of vitamin K1 was found on neurons from not only cortex, but also hippocampus, striatum, and septum, in addition to vitamin Kin, vitamin K 2 and K3 also showed the same effect on cortical neurons. These results suggest the potential role of the K vitamins on the maintenance of the survival of CNS neurons during the later stages of embryogenesis in vivo.
Phillips DJ, Greengard JS, Fernandez JA, Ribeiro M, Evatt BL, Griffin JH, Hooper WC. Protein S, an antithrombotic factor, is synthesized and released by neural tumor cells. J Neurochem. 1993;61:344–7.
Nunn JA, LePeillet E, Netto CA, Hodges H, Gray JA, Meldrum BS. Global ischaemia: hippocampal pathology and spatial deficits in the water maze. Behav Brain Res. 1994 May 30:62(1):41.-54.
Spatial deficits were assessed in male Wistar rats which had undergone 4 vessel occlusion for 5, 10, 15 or 30 min. Relationships between the extent of brain damage, the duration of 4-vessel occlusion, and the behavioral impairment consequent upon ischaemia were investigated. Starting 13–18 days after occlusion, rats were trained to find a hidden platform in a Morris water maze. All ischaemic groups were impaired on some performance indices relative to controls, in both acquisition and retention of the platform location. Increasing the duration of ischaemia increased behavioural deficits on some measures, but there was no clear-cut evidence that longer durations of ischaemia resulted in increased behavioural impairments. Histological assessment, at two coronal levels in hippocampus and four coronal levels in cortex and striatum, revealed CA1 cell loss in all ischaemic groups, which varied between 10–100% across the range of durations employed. CA1 cell loss increased as both a linear and quadratic function of increasing the duration of ischaemia. In rats subjected to 5–15 min ischaemia, cell loss was almost exclusively confined to the CA1 area. In rats subjected to 30 min ischaemia there was additional, variable damage in hippocampal areas CA2, 3 and 4, substantial cell loss in the striatum (50–70%) and some neuronal damage in the cortex (largely in layer III).
Thijssen HH, Drittij-Reijnders MJ. Vitamin K distribution in rat tissues: dietary phylloquinone is a source of tissue menaquinone-4. Br J Nutr. 1994;72:415–25.
The present study was undertaken to determine whether there is selective tissue distribution of vitamin K in the rat and whether this distribution mirrors the distribution of tissue vitamin K metabolism. The effects of feeding a vitamin K-free diet followed by resupplementation with phylloquinone (K1) were studied. K1 was recovered in all tissues. In K1-supplemented rats, most tissues accumulated K1 relative to plasma K1 with the highest levels in liver, heart, bone, and cartilaginous tissue (sternum). Low K1 levels were found in the brain. In the K1-free rats, relatively high K1 levels were still found in heart, pancreas, bone and sternum. Surprisingly, menaquinone-4 (MK-4) was detected in all tissues, with low levels in plasma and liver, and much higher levels in pancreas, salivary gland and sternum. MK-4 levels exceeded K1 levels in brain, pancreas, salivary gland and sternum. Supplementation with K1, orally and by intravenous infusion, caused MK-4 levels to rise. The results indicate that: (1) there is selective tissue distribution of K1 and MK-4, (2) dietary K1 is a source of MK-4.
He X, Shen L, Bjartell A, Dahlback B. The gene encoding vitamin K dependent anticoagulant protein S is expressed in multiple rabbit organs as demonstrated by northern blotting, in situ hybridization, and immunohistochemistry. J Histochem Cytochem. 1995;43:85–96. 50.
Jamison CS, Mcdowell SA, Marlar RA, Degen, SJ. Developmental expression of protein C and protein S in the rat. Thromb Res. 1995 Jun 1;78(5):407-19.
In order to better understand the expression of the Protein C/Protein S anticoagulant system, we have isolated and characterized cDNAs coding for rat Protein C and Protein S. From these results, it is evident that the expression of Protein C mRNA is similar to that of other vitamin K-dependent proteins. The expression of Protein S mRNA, however, is surprisingly complex and may include alternative splicing of mRNA to generate the various sizes evident on Northern analysis.
Ohashi K, Nagata K, Toshima J, Nakano T, Arita H, Tsuda H, et al. Stimulation of sky receptor tyrosine kinase by the product of growth arrest-specific gene 6. J Biol Chem. 1995;270: 22681–22684.
Reddi K, Henderson B, Meghji S, Wilson M, Poole S, Hopper C, Harris M, Hodges SJ. Interleukin 6 production by lipopolysaccharidestimulated human fibroblasts is potently inhibited by naphthoquinone (vitamin K) compounds. Cytokine. 1995;7:287–90.
This study examined the effect of various naphthoquinones on the production of interleukin 6 (IL-6) by lipopolysaccharide-stimulated human gingival fibroblasts. Compounds examined in this study include: phylloquinone (K1), menaquinone-4 (K2), menadione (K3), 2,3-dimethoxy-1,4-naphthoquinone (DMK) and a synthetic product of vitamin K catabolism, 2-methyl, 3-(2′methyl)-hexanoic acid-1,4-naphthoquinone (KCAT). All of these compounds are capable of inhibiting IL-6 production with a rank order of potency: KCAT>K3>DMK>K2>K1.
Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–70.
We report the identification of ligands for Tyro 3 (alternatively called Sky, rse, brt, or tif) and Axl (alternatively, Ark or UFO), members of a previously orphan family of receptor-like tyrosine kinases. These ligands correspond to protein S and Gas6. Our results are reminiscent of recent findings that the procoagulant thrombin, also binds and activates intracellular signaling via a G protein-coupled cell surface receptor. Proteases and protease regulators that also activate specific cell surface receptors may serve to integrate coagulation with associated cellular responses required for tissue repair and growth, as well as to coordinate protease cascades and associated cellular responses in other systems, such as those involved in growth and remodeling of the nervous system.
Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373:623–6.
Our results indicate that a vitamin-K-dependent protein known previously as a growth-arrest-specific gene is a ligand for Axl, a receptor tyrosine kinase. This is the first identification of a ligand for a receptor of the Axl family, and may aid the search for ligands for other Axl-related receptors. The initial identification of Gas6 as a protein expressed in response to growth arrest suggested that it may function as a negative regulator of cell proliferation. The potential role of Gas6 and Axl in cell proliferation is being further explored.
Wang, J.-Z., Gong, C.-X., Zaidi, T., Grundke-Iqbal, I. & Iqbal, K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995;270:4854–4860.
Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 1996;92:197–201.
Fridell Y-W, Jin Y, Quilliam LA, Burchert A, McCloskey P, Spizz G, et al. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 1996;16:135-145.
Goruppi S, Ruaro E, Schneider C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3Tc fibroblasts. Oncogene. 1996;12:471-80.
Reversible growth arrest has been characterized for enhanced expression of a set of genes called gas (growth arrest specific). Gas6 product (Gas6) is a secreted protein that was identified as the ligand for the tyrosine kinase receptor Axl. Here we report that Gas6 is able to induce cell cycle division entry in serum starved NIH3T3 cells. This mitogenic activity of Gas6 strictly correlates with its ability to interact with NIH3T3 endogenous Axl receptor. Finally we present evidences indicating that Gas6 is able to protect serum starved NIH3T3 cells from cell death by apoptosis as induced by complete growth factor depletion. The reported survival activity seems to be independent of Gas6 mitogenic activity, thus implicating a double and separable activity for Gas6 during growth arrest.
Gottfries, C. G., Karlsson, I. & Svennerholm, L. (1996) Membrane components separate early-onset Alzheimer's disease from senile dementia of the Alzheimer type. Int. Psychoger. 1996;8:365–372.
Brain tissue from 12 subjects with pure Alzheimer's disease (AD) and 21 subjects with senile dementia of the Alzheimer type (SDAT) was investigated for membrane lipids and compared with that in age-matched controls. In brain tissue from the patients with AD, phospholipids were significantly decreased compared with that from SDAT patients and controls, cholesterol was reduced compared with that in controls, and gangliosides were significantly reduced in all gray-matter areas investigated compared with those in both SDAT subjects and controls. The findings indicate synapse degeneration as an important pathogenetic factor in AD. This disorder should be separated from SDAT, in which white-matter degeneration appears to be more prominent.
Li R-H, Chen J, Hammonds G, Phillips H, Armanini M, Wood P, et al. Identification of Gas6 as a Growth Factor for Human Schwann Cells. J. Neurosci. 1996 Mar;16(6):2012-2019.
Schwann cells are one of the principal components of the peripheral nervous system. They play a crucial role in nerve regeneration and can be used clinically in the repair of injured nerves. We find that Gas6, a ligand for the Axl and Rse/Tyro3 receptor protein tyrosine kinase family, stimulates human Schwann ceil growth, increasing both cell number and thymidine incorporation. Gas6 has synergistic effects with the other known human Schwann cell mitogens, heregulin/glial growth factor and forskolin. A combination of Gas6 with heregulin and forskolin, on a defined background, supports maximal Schwann cell proliferation, while preserving the typical Schwann cell morphology and expression of the Schwann cell markers S-100, glial fibrillary acidic protein, and low-affinity nerve growth factor receptor. Gas6 mRNA is present in both spinal motor neurons and large neurons of the dorsal root ganglia, and neural injury has been reported to upregulate Rse/Axl in the Schwann cell. This is the first demonstration of a potentially important biological role for the human GasG/RseAxl system.
Mark MR, Chen J, Hammonds RG, Sadick M, Godowsk PJ. Characterization of Gas6, a member of the superfamily of G domain-containing proteins, as a ligand for Rse and Axl. J Biol Chem. 1996;271:9785–9789.
Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022-30027.
Rogers J, Webster S., Lue L-F, Brachova L, Civin WH, Emmerling M, et al. Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging. 1996;17:681–686.
The present review addresses these issues by showing that 1) inflammatory molecules and mechanisms are uniquely present or significantly elevated in the AD brain, 2) inflammation may be a necessary component of AD pathogenesis, 3) inflammation may be sufficient to cause AD neurodegeneration, and 4) retrospective and direct clinical trials suggest a therapeutic benefit of conventional antiinflammatory medications in slowing the progress or even delaying the onset of AD.
Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996; 271:18295–18298.
Alzheimer's disease (AD) is the most common progressive neurodegenerative disease known to humankind. It is characterized by brain atrophy, extracellular amyloid plaques, and intracellular neurofibril tangles. β-amyloid cascade is considered the major causative player in AD. Tyro3 receptor belongs to the TAM receptor subfamily of receptor protein tyrosine kinases (RPTKs). It is specifically expressed in the neurons of the neocortex and hippocampus. In this study, we established a cell model stably expressing APPswe mutants and producing Aβ. We found that overexpression of Tyro3 receptor in the cell model significantly decreased Aβ generation and also down-regulated the expression of β-site amyloid precursor protein cleaving enzyme (BACE1). However, the effects of Tyro3 were inhibited by its natural ligand, Gas6, in a concentration-dependent manner. In order to confirm the role of Tyro3 in the progression of AD development, we generated an AD transgenic mouse model accompanied by Tyro3 knockdown. We observed a significant increase in the number of amyloid plaques in the hippocampus in the mouse model. More plaque-associated clusters of astroglia were also detected. The present study may help researchers determine the role of Tyro3 receptor in the neuropathology of AD.
Sundaram K.S., Engelke J.A., Foley A.L., Suttie J., Lev M. Vitamin K Status Influences Brain Sulfatide Metabolism in Young Mice and Rats. J Nutr. 1996;126:2746–2751.
Administration of the vitamin K antagonist warfarin has previously been shown to decrease brain sulfatide concentrations and decrease brain galactocerebroside sulfotransferase (GST) activity in young mice. A dietary deficiency of vitamin K has now been shown to decrease (P < 0.01) brain sulfatide concentrations of 30-d-old mice significantly (by 21%). Male 21-d-old rats fed an excess of vitamin K for 7 or 14 days had 26 and 31% (P < 0.05) greater GST activity and 15 and 18% (P < 0.05) greater brain sulfatide concentrations, respectively, than controls fed a vitamin K-deficient diet. The vitamin K response was observed when either phylloquinone or menaquinone-4 was fed as a source of the vitamin. These data suggest that in addition to its recognized role in Gla synthesis, vitamin K status is important in the maintenance of normal complex lipid sulfatide metabolism in young rats and mice.
Thijssen HHW, Drittij-Reijnders MJ. Vitamin K status in human tissues: tissue-specific accumulation of phylloquinone and menaquinone-4. Brit. J Nutr. 1996;75:121-127.
We measured the vitamin K status in postmortem human tissues (brain, heart, kidney, liver, lung, pancreas) to see if there is a tissue-specific distribution pattern. Phylloquinone (K,) was recovered in all tissues with relatively high levels in liver, heart and pancreas; low levels were found in brain, kidney and lung. Menaquinone (MK-4) was recovered from most of the tissues; its levels exceeded the K levels in brain and kidney and equaled K, in pancreas. Liver, heart and lung were low in MK-4. The higher menaquinones, MK-6-11, were recovered in the liver samples, traces of MK-6-9 were found in some of the heart and pancreas samples. The results show that in man there are tissue-specific, vitamin-K distribution patterns comparable to those in the rat. Furthermore, the accumulation of vitamin K in heart, brain and pancreas suggests a hitherto unrecognized physiological function of this vitamin.
Turley SD, Burns DK, Rosenfeld CR, Dietschy JM. Brain does not utilize low density lipoprotein-cholesterol during fetal and neonatal development in the sheep. J. Lipid Res. 1996;37:1953–1961.
Herr DR, Fyrst H, Phan V, Heinecke K, Georges R, Harris GL. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Develop. 1997;130:2443-2453.
Leibson, C. L., Rocca, W. A., Hanson, V. A., Cha, R., Kokmen, E., O'Brien, P. C., et al. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann. N. Y. Acad. Sci. 1997;826:422–427.
Of the 1,455 cases of Adult Onset Diabetes Mellitus (AODM) followed for 9,981 person-years, 101 developed dementia, including 77 who met criteria for Alzheimer's disease. Persons with AODM exhibited significantly increased risk of all dementia. Risk of Alzheimer's disease was also elevated (for men, RR = 2.27, 95% Cl 1.55-3.31; for women, RR = 1.37, 95% Cl 0.94-2.01). These findings emphasize the importance of AODM prevention and prompt additional investigation of the relation between AODM and dementia.
McCloskey P, Fridell Y-W, Attar E, Jin Y, Varnum B, Liu ET. Gas6 mediates adhesion of cells expressing the receptor Tyrosine Kinase Asl. J Biol Chem. 1997;272(37):P23285-2329.
Lamon-Fava S, Sadowski JA, Davidson KW, O’Brien ME, McNamara JR, Schaefer EJ. Plasma lipoproteins as carriers of phylloquinone (vitamin K1) in humans. Am J Clin Nutr. 1998 Jun;67(6):1226-31.
The purpose of this study was to characterize the absorption and transport of phylloquinone (vitamin K1) by plasma lipoproteins. Twenty-six healthy subjects (11 men and 15 women) aged 20-78 y received phylloquinone in the amount of either 1.43 or 50 microg/kg body wt orally with a fat-rich meal containing 1.0 g/kg body wt of fat, carbohydrate, and protein and 7.0 mg cholesterol/kg body wt. Blood was obtained at baseline (0 hour) and 3, 6, 9, and 12 hours after the meal for the measurement of plasma lipid and phylloquinone concentrations in plasma and lipoprotein subfractions. In both groups of subjects, triacylglycerol concentrations peaked after 3 h in plasma and in the triacylglycerol-rich lipoprotein fraction, composed of chylomicrons and VLDLs. Plasma phylloquinone concentrations peaked at 6 hours. At baseline and during the postprandial phase, > 53% of plasma phylloquinone was carried by the triacylglycerol-rich lipoprotein fraction. In 9 of the 11 subjects supplemented with 50 microg phylloquinone/kg, plasma lipoproteins were isolated by sequential ultracentrifugation. In these subjects the fraction of plasma phylloquinone carried by LDLs and by HDLs increased progressively from 3% and 4% at 3 h to 14% and 11% at 12 h, respectively. Our data indicate that whereas triacylglycerol-rich lipoproteins are the major carriers of phylloquinone, LDL and HDL may carry small fractions of this vitamin.
Priefer BA, Robbins J. Eating changes in mild-stage Alzheimer’s disease: a pilot study. Dysphagia. 1997 Fall;12(4):212-21.
Eating impairment is well documented in the late stage of Alzheimer's disease (AD) but when these eating changes actually begin in the disease process is not known. Eating was defined as consisting of two components, self-feeding and swallowing. Self-feeding and swallowing of healthy elderly were compared with a group of individuals with mild AD. AD subjects received significantly more partner-initiated cues or direct assistance than controls. In addition, subject-initiated cued behaviors occurred more frequently in the AD group. AD subjects demonstrated significantly prolonged swallow durations for the oral transit duration (cookie), pharyngeal response duration (liquid), and total swallow duration (liquid). This pilot study suggests that self-feeding and swallowing changes may occur early in the course of AD.
Ueda K, Shinohara S, Yagami T, Kawasaki K. Amyloid beta protein potentiates Ca2+ influx through-Type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997 Jan;68(1):265-71.
Ueda K, Yagami T, Asakura k, Kawasaki K. Chlorpromazine reduces toxicity and Ca2+ uptake induced by amyloid β protein (25-35) in vitro. Brain Res. 1997 Feb 14;748(1-2):184-8.
Vervoort L.M.T., Ronden J.E., Thijssen H.H.W. The potent antioxidant activity of the vitamin K cycle in microsomal lipid peroxidation. Biochem. Pharmacol. 1997;54:871–876.
The results show that the vitamin K cycle could act as a potent antioxidant, that the active species in all probability is vitamin K-hydroquinone, and that the primary reaction product is the semiquinone. The results also show that the reaction product is processed in the vitamin K cycle to regenerate vitamin K-hydroquinone. Warfarin (5 μM) and chloro-vitamin K (50 μM), inhibitors of vitamin K epoxide reductase and γ-glutamylcarboxylase, respectively, were able to completely abolish the antioxidant effect.
Ariga T, Jarvis WD, Yu RK. Role of sphingolipid-mediated cell death in neurodegenerative diseases. J Lipid Res. 1998;39:1-16.
The origins of premature brain aging and chronic disease progression are associated with atherogenic diets and sedentary lifestyles in Western communities. Interests in brain aging that involves non alcoholic fatty liver disease (NAFLD), the global stroke epidemic and neurodegeneration have become the focus of nutritional research. Western diets high in fat induce hyperlipidemia, insulin resistance and other hormonal imbalances that are linked to alterations in brain calcium and lipid metabolism with susceptibility to various chronic diseases such as stroke. Nutrition and food science research identifies dietary components and lipids to prevent hyperlipidemia and calcium dyshomeostasis connected to neuroendocrine disease by maintaining astrocyte-neuron interactions and reversing hormonal imbalances that are closely associated with NAFLD, stroke and Alzheimer’s disease (AD) in global populations.
Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn A-M, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998 Nov;4:1313-17.
The genesis of new cells, including neurons, in the adult human brain has not yet been demonstrated. This study was undertaken to investigate whether neurogenesis occurs in the adult human brain, in regions previously identified as neurogenic in adult rodents and monkeys. Human brain tissue was obtained postmortem from patients who had been treated with the thymidine analog, bromodeoxyuridine (BrdU), that labels DNA during the S phase. Using immunofluorescent labeling, we demonstrate that new neurons, are generated from dividing progenitor cells in the dentate gyrus of adult humans. Our results further indicate that the human hippocampus retains its ability to generate neurons throughout life.
Davidson RT, Foley AL, Engelke JA, Suttie JW. Conversion of dietary phylloquinone to tissue menaquinone-4 in rats is not dependent on gut bacterial. J Nutr. 1998 Feb;128(2):220-3.
The ability of male rats to accumulate menaquinone-4 (MK-4) in tissues when fed a vitamin K-deficient diet supplemented with intraperitoneal phylloquinone (K) as the sole source of vitamin K for 14 days was assessed. In both conventionally housed controls and gnotobiotic rats, supplementation with the equivalent of 1500 microg vitamin K/kg diet increased tissue MK-4 concentrations above those of controls fed a vitamin K-deficient diet. MK-4 concentrations were approximately 5 ng/g in liver, 14 ng/g in heart, 17 ng/g in kidney, 50 ng/g in brain and 250 ng/g in mandibular salivary glands of gnotobiotic rats. MK-4 concentrations in conventionally housed rats were higher than in gnotobiotic rats in heart, brain and kidney but lower in salivary gland. Cultures of a kidney-derived cell line (293) converted K to the expoxide of MK-4 in a manner that was dependent on both time of incubation and concentration of vitamin K in the media. A liver-derived cell line (H-35) was less active in carrying out this conversion. These data offer conclusive proof that the tissue-specific formation of MK-4 from K is a metabolic transformation that does not require bacterial transformation to menadione as an intermediate in the process.
Snipes G, Suter U. 1998. Cholesterol and Myelin. In: Bittman R, ed. Cholesterol. New York: Plenum Press.
In this review, we will summarize progress in our understanding of the role of cholesterol in the CNS and PNS with a particular emphasis on myelin.
Allen MP, Zeng C, Schneider K, Xiong X, Meintzer MK, Bellosta P, Basilico C, Varnum B, Heidenreich KA, Wierman ME. Growth arrest specific gene 6 (Gas6)/adhesion related kinase (Ark) signaling promotes gonadotropin-releasing hormone neuronal survival via extracellular signalregulated kinase (ERK) and Akt. Mol Endocrinol. 1999;13:191–201.
Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhoj P, Pedersen BK. A high plasma concentration of TNF- X is associated with dementia in centenarians. J. of Gerontology: Medical Sciences. 1999;54A(7):M357-364.
Inflammatory mechanisms and immune activation have been hypothesized to play a role In the pathogenesis of age-associated diseases such as dementia and atherosclerosis. The purpose of this study was to evaluate the plasma concentration of tumor necrosis factor(1NF)-ex in a large cohort of centenarians and to look for its possible associations with cognitive function, atherosclerosis ,and general health status. We found that the concentration of TNF-ex was significantly increased in 126 centenarians compared to younger control groups, and a high concentration of TNF-a was associated with both Alzheimer's disease and generalized atherosclerosis in the centenarians. The concentration of TNF-ex was positively correlated with the concentrations of plasma IL-6,s TNFR-II, and CRP. This study demonstrates that, even in apparently healthy subjects, age-associated immune activation indicated by raised levels of pro-inflammatory cytokines may reflect age-associated pathological processes that develop over decades.
Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999 Sep;24(9):329-32.
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 1999;2:260–265.
Thousands of hippocampal neurons are born in adulthood, suggesting that new cells could be important for hippocampal function. To determine whether hippocampus-dependent learning affects adult-generated neurons, we examined the fate of new cells labeled with the thymidine analog bromodeoxyuridine following specific behavioral tasks. Here we report that the number of adult-generated neurons doubles in the rat dentate gyrus in response to training on associative learning tasks that require the hippocampus. In contrast, training on associative learning tasks that do not require the hippocampus did not alter the number of new cells. These findings indicate that adult-generated hippocampal neurons are specifically affected by, and potentially involved in, associative memory formation.
Huber AM, Davidson KW, O’Brien-Morse ME, Sadowski JA. Tissue phylloquinone and menaquinones in rats are affected by age and gener. J Nutr. 1999;129:1039-44.
Lim DA, Alvarez-Buylla A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci USA. 1999 Jun 22;96(13):7526-31.
Neurogenesis continues in the mammalian subventricular zone (SVZ) throughout life. However, the signaling and cell-cell interactions required for adult SVZ neurogenesis are not known. In vivo, migratory neuroblasts (type A cells) and putative precursors (type C cells) are in intimate contact with astrocytes (type B cells). Type B cells also contact each other. We reconstituted SVZ cell-cell interactions in a culture system free of serum or exogenous growth factors. Culturing dissociated postnatal or adult SVZ cells on astrocyte monolayers-but not other substrates-supported extensive neurogenesis similar to that observed in vivo. SVZ precursors proliferated rapidly on astrocytes to form colonies containing up to 100 type A neuroblasts. By fractionating the SVZ cell dissociates with differential adhesion to immobilized polylysine, we show that neuronal colony-forming precursors were concentrated in a fraction enriched for type B and C cells. Pure type A cells could migrate in chains but did not give rise to neuronal colonies. Because astrocyte-conditioned medium alone was not sufficient to support SVZ neurogenesis, direct cell-cell contact between astrocytes and SVZ neuronal precursors may be necessary for the production of type A cells.
Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999 Apr 22;398(6729):723-8.
We have generated and analysed null mutations in the mouse genes encoding three structurally related receptors with tyrosine kinase activity: Tyro 3, Axl, and Mer. Mice lacking any single receptor, or any combination of two receptors, are viable and fertile, but male animals that lack all three receptors produce no mature sperm, owing to the progressive death of differentiating germ cells. This degenerative phenotype appears to result from a failure of the tropic support that is normally provided by Sertoli cells of the seminiferous tubules, whose function depends on testosterone and additional factors produced by Leydig cells. Tyro 3, Axl and Mer are all normally expressed by Sertoli cells during postnatal development, whereas their ligands, Gas6 and protein S, are produced by Leydig cells before sexual maturity, and by both Leydig and Sertoli cells thereafter. Here we show that the concerted activation of Tyro 3, Axl and Mer in Sertoli cells is critical to the role that these cells play as nurturers of developing germ cells. Additional observations indicate that these receptors may also be essential for the tropic maintenance of diverse cell types in the mature nervous, immune and reproductive systems.
Prieto AL, Weber JL, Tracy S, Heeb MJ, Lai C. Gas6, a ligand for the receptor protein-tyrosine kinase Tyro-3, is widely expressed in the central nervous system. Brain Res. 1999;816:646–61.
Gas6 (growth arrest specific gene-6) is a ligand for members of the Axl subfamily of receptor protein-tyrosine kinases. One of these receptors, Tyro-3, is widely expressed in the central nervous system. We have used biochemical and histological techniques, to determine the expression patterns of Gas6 mRNA and protein during development. Gas6 is widely expressed in the rat central nervous system (CNS) beginning at late embryonic stages and its levels remain high in the adult. At embryonic day 14 it is detected in the heart, blood vessels, testes, choroid plexus, and in the ventral spinal cord. In the adult, Gas6 is expressed in the cerebral cortex, (predominantly in layer V), the piriform cortex, and the hippocampus (areas CA1, CA3 and the dentate gyrus). It is also expressed in thalamic and hypothalamic structures, the midbrain, and in a subset of motor and trigeminal nuclei. In the cerebellum, it is expressed in Purkinje neurons and deep cerebellar nuclei. Protein S, a protein related to Gas6, is only detected at low levels in the CNS. The spatial and temporal profiles of Gas6 expression suggest that it could potentially serve as the physiologically relevant ligand for Tyro-3 in the postnatal rat nervous system.
Reisberg B, Franssen EH, Hasan SM, Monteiro I, Boksay I, Souren LE, et al. Retrogenesis: clinical, physiologic, and pathologic mechanisms in brain aging, Alzheimer's and other dementing processes. Eur Arch Psychiatry Clin Neurosci. 1999;249(Suppl 3):28–36.
Schurgers LJ, Geleijnse JM, Grobbee DE, Pols HAP, Hofman A, Witteman JCB, et al. Nutritional intake of vitamins K1 (phylloquinone) and K2 (menaquinone) in the Netherlands. J Nutr Environ Med. 1999;9:115-122.
Vitamin K plays a key role in the hepatic synthesis of blood clotting factors. Recently, other tissues (bone, vessel wall) were shown to produce vitamin K-dependent proteins not involved in blood coagulation. Multiple forms of vitamin K have been found in human food: phylloquinone (K1) and various menaquinones. A recommended dietary allowance (RDA) has only been defined for K1, and its value is exclusively based on blood clotting data. We have prepared a provisional table of menaquinones in food, which has been used to calculate the total vitamin K intake in a well-defined cohort of the Dutch population. It is concluded that K1 is the major form of nutritional vitamin K, that total vitamin K intake is higher than in other populations described and that the correlation between vitamin K intake and serum concentration is poor. It is suggested that present RDA values be reconsidered and intakes comparable with those in the highest quartile of our study population are recommended.
Tsaioun K. Vitamin K-dependent proteins in the developing and aging nervous system. Nutr Revs. 1999 Aug;57(8):231-240.
Although the impact of warfarin on embryo development, with its neurologic and bone abnormalities, has been known for decades, the role of vitamin K in the brain has not been studied systematically. Recently, it was demonstrated that vitamin K-dependent carboxylase expression is temporally regulated in a tissue-specific manner with high expression in the nervous system during the early embryonic stages and with liver expression after birth and in adult animals. This finding, along with the discovery of wide distribution of the novel vitamin K-dependent growth factor, Gas6, in the central nervous system, provides compelling evidence of a biologic role of vitamin K during the development of the nervous system. In animals and bacteria, vitamin K was observed to influence the brain sulfatide concentration and the activity and synthesis of an important enzyme in volved in brain sphingolipids biosynthesis. Taken together, previous research results point to a possible role of vitamin K in the nervous system, especially during its development. Hence, the knowledge of the biologic role of vitamin K in the brain may be important for unveiling the mechanisms of normal and pathologic development and aging of the nervous system. The role of the vitamin K-dependent protein Gas6 in activation of signal transduction events in the brain in light of the age-related changes in the nervous system is also discussed.
2000
Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14 Suppl 1:S47-53.
Lesions of Alzheimer disease are associated with low-grade but sustained inflammatory responses. Activated microglia agglomerate in the center of senile plaques. Reactive astrocytes marginate the amyloid beta-protein (A beta) deposits and extend their processes toward the center of plaques. Both microglia and astrocytes are known to secrete a wide variety of molecules involved in inflammation and are potential sources of proinflammatory elements in the brain. Dystrophic neurites occur in senile plaques with such glial reactions, suggesting the relevance of inflammatory responses to the neuronal degeneration in Alzheimer disease. Activated glial cells are, therefore, targets of anti-inflammatory therapy of Alzheimer disease. However, evidence also indicates that these cells eliminate A beta from the brain. A beta is produced continuously in both the normal and the AD brain. Under normal conditions, A beta is removed successfully before it accumulates as extracellular amyloid fibrils. Even in Alzheimer disease, a large portion of A beta may be cleared from the brain with a small portion being left and deposited as neurotoxic senile plaques. Both in vivo and in vitro studies showed the effective uptake of A beta by microglia. Before clinical application, it must be determined whether the treatment that suppresses glial activation and inflammatory responses inhibits A beta removal by glial cells.
Benzakour O, Kanthou C. The anticoagulant factor, protein S, is produced by cultured human vascular smooth muscle cells and its expression is up-regulated by thrombin. Blood. 2000 Mar 15;95(6):2008-14.
The anticoagulant factor protein S is a secreted vitamin K-dependent gamma-carboxylated protein that is mainly made in the liver. Protein S is homologous to the growth arrest specific protein, Gas6, the expression of which is up-regulated in cultured fibroblasts upon serum withdrawal. We report here the synthesis and secretion of protein S by cultured human vascular smooth muscle cells (HVSMCs). Western blot analysis revealed that similar amounts of protein S are secreted by both growing and growth-arrested HVSMCs. HVSMC-derived protein S was found to be gamma-carboxylated as it was precipitated by barium citrate and was shown to possess protein C cofactor activity. Treatment with the vitamin K antagonist warfarin led to the accumulation of intracellular undercarboxylated protein S forms that were rapidly secreted upon the reintroduction of vitamin K. The evidence we provide for protein S secretion by cultured HVSMCs and its up-regulation by thrombin, together with earlier reports showing that protein S acts as a mitogen for these cells, suggests that, in addition to its known role in regulating blood clotting, protein S may also be an important autocrine factor in the pathophysiology of the vasculature.
Bolton-Smith C, Price RJG, Fenton ST, Harrington DJ, Shearer MJ. Compilation of a provisional UK database for the phylloquinone (vitamin K1) content of foods.
Br. J. Nutr. 2000;83:389-399.
Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–8.
Neural stem cells exist not only in the developing mammalian nervous system but also in the adult nervous system of all mammalian organisms, including humans. Neural stem cells can also be derived from more primitive embryonic stem cells. The location of the adult stem cells and the brain regions to which their progeny migrate in order to differentiate remain unresolved, although the number of viable locations is limited in the adult. The mechanisms that regulate endogenous stem cells are poorly understood. Potential uses of stem cells in repair include transplantation to repair missing cells and the activation of endogenous cells to provide "self-repair. " Before the full potential of neural stem cells can be realized, we need to learn what controls their proliferation, as well as the various pathways of differentiation available to their daughter cells.
Goodrum JF, Brown JC, Fowler KA, Bouldin TW. Axonal regeneration, but not myelination, is partially dependent on local cholesterol reutilization in regenerating nerve. J Neuropathol Exp Neurol. 2000;59:1002–10.
Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. 2000;127(3):411–417.
Prieto AL, Weber JL, Lai C. Expression of the receptor protein-tyrosine kinases Tyro-3, Axl, and Mer in the developing rat central nervou system. J Com Neur. 2000;42592):295-314.
Tyro-3, Axl, and Mer are three related receptor protein-tyrosine kinases (RPTKs) characterized by an extracellular domain exhibiting significant amino acid sequence similarity to neural cell adhesion molecules. The molecule Gas6 (for growth arrest-specific gene-6) has been shown to activate each of these receptors. Gas6 is expressed extensively in the central nervous system (CNS), suggesting that interactions between Gas6 and its receptors are likely to have physiologically relevant functions. To identify and localize the relevant Gas6/RPTK pairs, we have characterized the developmental expression of Tyro-3, Axl, and Mer in rat CNS. Throughout development, Tyro-3 was the most widely expressed of the three receptors in the CNS, with Axl and Mer detected in only a limited number of sites in the adult. Tyro-3 expression was low in the embryo and increased markedly during early postnatal stages, with a time course paralleling that of synaptogenesis. Axl and Mer were expressed at low but relatively constant levels throughout development. In the cerebellum, all three receptors were found in Purkinje cells, and Tyro-3 was also detected in both granule neurons and Bergmann glia. Insofar as Gas6 has been previously shown to also be expressed by Purkinje cells, it may be engaged in both autocrine and paracrine signaling. The three receptors were also detected in cerebellar white matter, primarily during myelination. In the cortex, Tyro-3 was expressed at high levels during postnatal development and in the adult. Beginning at P6 in the hippocampus, Tyro-3 was expressed at high levels in CA1 pyramidal neurons and at lower levels in CA3 and was not detected in dentate granule neurons. Axl and Mer were found in the molecular layer of the dentate gyrus and were absent from the pyramidal and dentate granule neurons. In that Gas6 is expressed throughout the pyramidal cell layer, it may activate these cells in both an autocrine and a paracrine manner. These studies provide initial clues for elucidating the cellular functions of the Axl subfamily members and suggest potential complex Gas6/RPTK as well as RPTK/RPTK signaling interactions in the mature and developing CNS.
Schurgers LJ, Vermeer C. Determination of phylloquinone and menaquinones in food. Effect of food matrix on circulating vitamin K concentrations. Haemostasis. 2000 Nov-Dec;30(6):298-307.
Fluctuations in international normalized ratio values are often ascribed to dietary changes in vitamin K intake. Here we present a database with vitamin K(1) and K(2) contents of a wide variety of food items. K(1) was mainly present in green vegetables and plant margarins, K(2) in meat, liver, butter, egg yolk, natto, cheese and curd cheese. To investigate the effect of the food matrix on vitamin K bioavailability, 6 healthy male volunteers consumed either a detergent-solubilized K(1) or a meal consisting 400 g of spinach K(1)) and 200 g of natto K2. The absorption of pure K(1) was faster than that of food-bound K vitamins (serum peak values at 4 h vs. 6 hours after ingestion). Moreover, circulating K(2) concentrations after the consumption of natto were about 10 times higher than those of K(1) after eating spinach. It is concluded that the contribution of K(2) vitamins (menaquinones) to the human vitamin K status is presently underestimated, and that their potential interference with oral anticoagulant treatment needs to be investigated.
Tsaioun KI, Denisova NA, Obin M, Joseph J. Novel growth factor Gas6, phosphatidylserine and their age-related changes in the rat brain. Neurosci Res Commun. 2000;26:113–22.
Allison, A.C. The possible role of vitamin K deficiency in the pathogenesis of Alzheimer’s disease and in augmenting brain damage associated with cardiovascular disease. Med. Hypotheses 2001, 57, 151–155.
The incidence of Alzheimer's disease (AD) increases with age and in carriers of the apolipoprotein E4 genotype. A relative deficiency of vitamin K, affecting the extrahepatic functions of the vitamin, is common in ageing men and women. The concentration of vitamin K is lower in the circulating blood of APOE4 carriers than in that of persons with other APOE genotypes. Evidence is accumulating that vitamin K has important functions in the brain, including the regulation of sulfotransferase activity and the activity of a growth factor/tyrosine kinase receptor (Gas 6/Axl). The hypothesis is now proposed that vitamin K deficiency contributes to the pathogenesis of AD and that vitamin K supplementation may have a beneficial effect in preventing or treating the disease. Vitamin K may also reduce neuronal damage associated with cardiovascular disease.
Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927.
Cutler RG, Mattson MP. Sphingomyelin and ceramide as regulators of development and lifespan. Mech Ageing Dev. 2001 Jul 15;122(9):895-908.
Sphingomyelin (SM) is a prominent phospholipid component of cell membranes that has evolved diverse functions in cells beyond its role in membrane structural organization. Cleavage of SM by acid or neutral sphingomyelinase results in the liberation of ceramide, an intracellular messenger that regulates the activities of an array of kinases, phosphatases and transcription factors. Signals that activate sphingomyelinases range from growth factors and cytokines, to neurotransmitters, hormones and reactive oxygen species. Studies of experimental cell culture and animal models, and of patients with inherited defects in sphingomyelin metabolism suggest important roles for SM-ceramide signaling in the regulation of cell proliferation, differentiation and survival. At low concentrations SM and ceramide can stimulate cell proliferation and survival, whereas higher levels can induce cell dysfunction or death. Analyses of development and aging suggest a major role for SM metabolism in regulating development rate and lifespan. Several factors that alter the metabolism of sphingolipids, including oxidative and metabolic stress, also increase risk and progression of age-related diseases. In addition, recent findings have linked alterations in SM metabolism to the pathogenesis of several age-related diseases including cancers and neurodegenerative disorders. The emerging data suggest the possibility that dietary and pharmacological manipulations of SM metabolism might prove effective in extending lifespan and treating various age-related diseases.
Gomes FC, Spohr TC, Martinez R, Moura Neto V. Cross-talk between neurons and glia: highlights on soluble factors. Braz J Med Biol Res. 2001 May;34(5):611-20.
The development of the nervous system is guided by a balanced action between intrinsic factors represented by the genetic program and epigenetic factors characterized by cell-cell interactions which neural cells might perform throughout nervous system morphogenesis. Highly relevant among them are neuron-glia interactions. In this review, we will focus our attention on recent advances in the understanding of the role of glial and neuronal trophic factors in nervous system development. We will argue that the functional architecture of the brain depends on an intimate neuron-glia partnership.
Ohanian J, Ohanian V. Sphingolipids in mammalian cell signalling. Cell Mol Life Sci. 2002 Dec;58(14):2053-68.
Sphingolipids and their metabolites, ceramide, sphingosine and sphingosine-1-phosphate, are involved in a variety of cellular processes including differentiation, cellular senescence, apoptosis and proliferation. Ceramide is the main second messenger, and is produced by sphingomyelinase-induced hydrolysis of sphingomyelin and by de novo synthesis. Sphingomyelin in the plasma membrane is located primarily in the outer (extracellular) leaflet of the bilayer, whilst sphingomyelinases are found at the inner (cytosolic) face and within lysosomes/endosomes. Glycosphingolipids and sphingomyelin together with cholesterol are major components of specialised membrane microdomains known as lipid rafts, which are involved in receptor aggregation and immune responses. Many signalling molecules, for example Src family tyrosine kinases and glycosylinositolphosphate-anchored proteins, are associated with rafts, and disruption of these domains affects cellular responses such as apoptosis. The importance of sphingolipid signalling in cardiovascular development has been reinforced by recent reports implicating EDG receptors in the regulation of embryonic cardiac and vascular morphogenesis.
Rivière S, Gillette-Guyonnet S, Voisin T, Reynish E, Andrieu S, Lauque S, et al. A nutritional education program could prevent weight loss and slow cognitive decline in Alzheimer’s disease. J Nutrl Health Aging. 2001;5(4):295-9.
Weight loss is a common problem in patients with Alzheimer's Disease (AD). It is a predictive factor of mortality and it decreases patients' and caregivers' quality of life. This study looked at whether a nutritional education program can prevent weight loss in AD patients. 151 AD patients and their caregivers were enrolled to follow the intervention and 74 AD patients and their caregivers constituted a control group. Caregivers in the intervention group followed 9 nutritional sessions of one hour each, over one year. Caregivers in the control group didn't follow any sessions but were offered advice provided in a normal follow-up. The results suggest that a nutritional educational program intended for caregivers of AD patients could have a positive effect on patient’s weight and cognitive function.
Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Proceedings of the Nutrition Society. 2001;60:135-143.
Certain dietary risk factors for physical ill health are also risk factors for depression and cognitive impairment. Although cholesterol lowering has been suggested to increase vulnerability to depression, there is better support for an alternative hypothesis that intake of n-3 long-chain polyunsaturated fatty acids can affect mood (and aggression). Possible mechanisms for such effects include modification of neuronal cell membrane fluidity and consequent impact on neurotransmitter function. Stronger evidence exists concerning a role for diet in influencing cognitive impairment and cognitive decline in older age, in particular through its impact on vascular disease. For example, cognitive impairment is associated with atherosclerosis, type 2 diabetes and hypertension, and findings from a broad range of studies show significant relationships between cognitive function and intakes of various nutrients, including long-chain polyunsaturated fatty acids, antioxidant vitamins, and folate and vitamin B12. Further support is provided by data on nutrient status and cognitive function. Almost all this evidence, however, comes from epidemiological and correlational studies. Given the problem of separating cause and effect, greater emphasis should now be placed on conducting intervention studies. An efficient approach to this problem could be to include assessments of mood and cognitive function as outcome measures in studies designed primarily to investigate the impact of dietary interventions on markers of physical health.
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS & Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207– 211.
Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376.
The vertebrate brain continues to produce new neurons throughout life. In the rat hippocampus, several thousand are produced each day, many of which die within weeks. Associative learning can enhance their survival; however, until now it was unknown whether new neurons are involved in memory formation. Here we show that a substantial reduction in the number of newly generated neurons in the adult rat impairs hippocampal-dependent trace conditioning, a task in which an animal must associate stimuli that are separated in time. A similar reduction did not affect learning when the same stimuli are not separated in time, a task that is hippocampal-independent. The reduction in neurogenesis did not induce death of mature hippocampal neurons or permanently alter neurophysiological properties of the CA1 region, such as long-term potentiation. Moreover, recovery of cell production was associated with the ability to acquire trace memories. These results indicate that newly generated neurons in the adult are not only affected by the formation of a hippocampal-dependent memory, but also participate in it.
Allen MP, Linseman DA, Udo H, Xu M, Schaack JB, Varnum B, Kandel ER, Heidenreich KA, Wierman ME. Novel mechanism for gonadotropin-releasing hormone neuronal migration involving Gas6/Ark signaling to p38 mitogen-activated protein kinase. Mol Cell Biol. 2002;22:599–613.
Burns M, Duff K. Cholesterol in Alzheimer’s disease and tauopathy. Ann NY Acad Sci. 2002;977:367-375.
Dobson CM. Getting out of shape. Nature. 2002 Aug 15;418(6899):729-30.
Funakoshi H, Yonemasu T, Nakano T, Matumoto K, Nakamura T. Identification of Gas6, a putative ligand for Sky and Axl receptor tyrosine kinases, as a novel neurotrophic factor for hippocampal neurons. J Neurosci Res. 2002;68:150–60.
Han X, Holtzman DM, McKeel Jr DW, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J. Neurochem. 2002;82:809-818.
The findings demonstrate that a marked decrease in sulfatides is associated with AD pathology even in subjects with very mild dementia and that these changes may be linked with early events in the pathological process of AD.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353-6.
It has been more than 10 years since it was first proposed that the neurodegeneration in Alzheimer's disease (AD) may be caused by deposition of amyloid beta-peptide (Abeta) in plaques in brain tissue. According to the amyloid hypothesis, accumulation of Abeta in the brain is the primary influence driving AD pathogenesis. The rest of the disease process, including formation of neurofibrillary tangles containing tau protein, is proposed to result from an imbalance between Abeta production and Abeta clearance.
Jellinger AK. Alzheimer disease and cerebral vascular pathology: an update. J Neural Transm (Vienna). 2002 May;109(5-6):813-36.
Recent epidemiological and clinico-pathologic data suggest overlaps between Alzheimer disease (AD) and cerebrovascular lesions that may magnify the effect of mild AD pathology and promote progression of cognitive decline or even may precede neuronal damage and dementia. Vascular pathology in the aging brain and in AD includes: 1. cerebral amyloid angiopathy 2. Microvascular changes with decreased density and structural abnormalities causing regional metabolic and blood-brain barrier dysfunctions with ensuing neuronal damage. In large autopsy series of demented aged subjects, around 80% show Alzheimer type pathology, 20-40% with additional, often minor vascular lesions, 7-10% "pure" vascular dementia, and 3-5% "mixed" dementia (combination of AD and vascular encephalopathy). AD cases with additional minor cerebrovascular lesions have significantly more frequent histories of hypertension or infarcts than "pure" AD patients. Vascular lesions in AD include cortical microinfarcts, subcortical lacunes, white matter lesions / leukoencephalopathy, small hemorrhages and corticosubcortical infarcts, while in mixed type dementia multiple larger or hemispheral infarcts are more frequent. Small infarcts in AD patients have no essential impact on global cognitive decline which mainly depends on the severity of Alzheimer pathology, but in early stage of AD they may influence and promote the development of dementia. Recent studies showed lower density of plaques and tangles in brains with cerebrovascular lesions, and similar severity of dementia was related to fewer AD lesions in brains with than in those without small vascular lesions. Further studies will help to elucidate the risk factors and impact of cerebrovascular lesions on the development and progression of dementia in AD.
Levade T, Malagarie-Cazenave S, Gouazé V, Ségui B, Tardy C, Betito S, et al. Ceramide in apoptosis: a revisited role. Neurochem Res. 2002 Aug;27(7-8).601-7.
The sphingolipid ceramide has recently emerged as a new transducer or modulator of apoptotic cell death. This function, however, has recently been challenged. Here, in the light of recent observations, the role of ceramide in apoptosis signaling is discussed.
Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002 Jun;3(6):423-32.
The conventional scheme of cortical formation shows that postmitotic neurons migrate away from the germinal ventricular zone to their positions in the developing cortex, guided by the processes of radial glial cells. However, recent studies indicate that different neuronal types adopt distinct modes of migration in the developing cortex. Here, we review evidence for two modes of radial movement: somal translocation, which is adopted by the early-generated neurons; and glia-guided locomotion, which is used predominantly by pyramidal cells. Cortical interneurons, which originate in the ventral telencephalon, use a third mode of migration. They migrate tangentially into the cortex, then seek the ventricular zone before moving radially to take up their positions in the cortical anlage.
Nakagawa S, Kim J-E, Lee R, Chen J, Fujioka T, Malberg J, et al. Localization of phosphorylated cAMP Response Element-Binding protein in immature neurons of adult hippocampus. J. Neurosci. 2002 Nov 15;22(22):9868-76.
Neurogenesis continues to occur in the adult hippocampus, although many of the newborn cells degenerate 1–2 weeks after birth. The number and survival of newborn cells are regulated by a variety of environmental stimuli, but very little is known about the intracellular signal transduction pathways that control adult neurogenesis. In the present study, we examine the expression of the phosphorylated cAMP response element-binding protein (pCREB) in immature neurons in adult hippocampus and the role of the cAMP cascade in the survival of new neurons. The results demonstrate that virtually all immature neurons, identified by triple immunohistochemistry for bromodeoxyuridine (BrdU) and polysialic acid-neural cell adhesion molecule (PSA-NCAM), are also positive for pCREB. In addition, upregulation of cAMP (via pharmacological inhibition of cAMP breakdown or by antidepressant treatment) increases the survival of BrdU-positive cells. A possible role for pCREB in the regulation of PSA-NCAM, a marker of immature neurons involved in neuronal remodeling and neurite outgrowth, is supported by cell culture studies demonstrating that the cAMP–CREB pathway regulates the expression of a rate-limiting enzyme responsible for the synthesis of PSA-NCAM. These findings indicate that the cAMP–CREB pathway regulates the survival, and possibly the differentiation and function, of newborn neurons.
Thane CW, Paul AA, Bates CJ, Bolton-Smith C, Prentice A, Shearer MJ. Intake and sources of phylloquinone (vitamin K1): variation with socio-demographic and lifestyle factors in a national sample of British elderly people. Br J Nutr. 2002;87:605-613.
Intake and sources of phylloquinone (vitamin K1) were examined according to socio-demographic and lifestyle factors in free-living British people aged 65 years and over, from the 1994–5 National Diet and Nutrition Survey. Of all the participants, 59 % had phylloquinone intakes below the current guideline for adequacy of 1 μg/kg body weight per d. Participants aged 85 years and over, formerly in manual occupations, or living in Scotland or in northern England reported lower phylloquinone intakes than their comparative groups. Overall, vegetables contributed 60 % of total phylloquinone intake, with cooked green vegetables providing around 28 % of the total. Dietary supplements contributed less than 0·5 % of phylloquinone intake. Participants living in northern England or in Scotland, in particular, derived less phylloquinone from vegetables than those living in southern England.
Tsang, C.K.; Kamei, Y. Novel effect of vitamin K1 (phylloquinone) and vitamin K2 (menaquinone) on promoting nerve growth factor-mediated neurite outgrowth from PC12D cells. Neurosci. Lett. 2002, 323, 9–12.
Yagami T, Ueda K, Asakura K, Sakaeda T, Nakazato H, Kuroda T, et al. Gas6 rescues cortical neurons from amyloid beta protein-induced apoptosis. Neuropharmacology. 2002;43:1289–96.
Gas6, a product of the growth-arrest-specific gene 6, protects neurons from serum deprivation-induced apoptosis. Neuronal apoptosis is also caused by amyloid β protein (Aβ), whose accumulation in the brain is a characteristic feature of Alzheimer’s disease. Aβ induces Ca2+ influx via L-type voltage-dependent calcium channels (L-VSCCs), leading to its neurotoxicity. In the present study, we investigated effects of Gas6 on Aβ-induced cell death in primary cultures of rat cortical neurons. Aβ caused neuronal cell death in a concentration- and time-dependent manner. Gas6 significantly prevented neurons from Aβ-induced cell death. Prior to cell death, Aβ increased influx of Ca2+ into neurons through L-VSCCs. Gas6 significantly inhibited the Aβ-induced Ca2+ influx. The inhibitor of L-VSCCs also suppressed Aβ-induced neuronal cell death. The present cortical cultures contained few non-neuronal cells, indicating that Gas6 affected the survival of neurons directly, but not indirectly via non-neuronal cells. In conclusion, we demonstrate that Gas6 rescues cortical neurons from Aβ-induced apoptosis. Furthermore, the present study indicates that inhibition of L-VSCC contributes to the neuroprotective effect of Gas6.
Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol. 2003;4(1):87–91.
Bartzokis G, Cummings JL, Sultzer D, Henderson VW, Nuechterlein KH, Mintz J. White matter structural integrity in healthy aging adults and patients with Alzheimer disease: a magnetic resonance imaging study. Arch Neurol. 2003;60:393–398.
Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74(7):857–62.
Analysis of all 15 case-control studies was significant (OR 1.58, 95% CI 1.21 to 2.06), indicating an excess history of head injury in those with Alzheimer's disease. The finding of Mortimer et al that head injury is a risk factor for Alzheimer's disease only in males was replicated. The excess risk of head injury in those with Alzheimer's disease is only found in males (males: OR 2.29, 95% CI 1.47 to 2.06; females: OR 0.91, 95% CI 0.56 to 1.47). This study provides support for an association between a history of previous head injury and the risk of developing Alzheimer's disease.
Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003 May;60(5):729-36.
Large numbers of neurofibrillary tangles (NFTs) and amyloid plaques are diagnostic markers for Alzheimer disease (AD), but lesser numbers of these lesions are also seen in nondemented elderly individuals. Much of the existing literature suggests that the NFTs of AD have a closer correlation with cognitive function than do amyloid plaques. Whether a similar relationship exists in normal aging and mild cognitive impairment (MCI), a condition that frequently reflects a preclinical stage of AD, remains unknown. This study looked at the distribution patterns of beta-amyloid plaques and NFTs and the association of these lesions with memory performance in nondemented individuals. Eight nondemented subjects who volunteered to receive annual neuropsychological testing and agreed to brain donation were studied. Five subjects showed no cognitive impairment, and 3 were diagnosed with MCI. The distribution of NFTs followed a rigorous and hierarchical pattern, but distribution of amyloid plaques varied among individuals. Subjects with MCI displayed higher NFT densities than did nonimpaired subjects. In addition, NFT density in the temporal lobe correlated with memory scores, whereas density of amyloid plaques did not. Neurofibrillary tangles are more numerous in medial temporal lobe regions associated with memory function and show a relationship to performance on memory tests in nondemented individuals. These results suggest that NFTs may constitute a pathological substrate for memory loss not only in AD but also in normal aging and MCI.
Hall MO, Agnew BJ, Abrams TA, Burgess BL. 2003. The phagocytosis of OS is mediated by the PI3-Kinase linked Tyrosine Kinase receptor, Mer, and is stimulated by Gas6. In: LaVail MM, Hollyfield JG, Anderson RE, Editors. Retinal Degenerations: Mechanisms and Experimental Therapy. New York: Kluwer Academic/Plenum Publishers.
Holm B, Söderhamn O. Factors associated with nutritional status in a group of people in an early stage of dementia. Clin Nutr. 2003 Aug;22(4):385-9.
Beside cognitive and behavioural problems, nutritional difficulties and weight loss are often observed at the beginning of dementia disease. This weight loss has been hard to explain. But previous research has shown that it is not related to increased energy expenditure but rather to deficiency in intake at the beginning of the disease. The aim of this study was to investigate possible factors that may be associated with nutritional status among people in an early stage of dementia. Fifty-nine individuals with perceived impaired memory were interviewed with three assessment instruments and a number of other structured questions. The results showed that predictors for nutritional status were: eating smaller portions, partly lost learned practices, and having a dry mouth. Caring actions for this patient group should focus on support at meals and maintaining oral health.
Lemke G., Lu Q. Macrophage regulation by Tyro 3 family receptors. Curr. Opin. Immunol. 2003;15:31–36.
Recent analyses demonstrate that the three receptor protein-tyrosine kinases of the Tyro 3 family function as central regulators of the activation state of macrophages. These studies, carried out in cells and tissues derived from single, double and triple mouse knockouts, have shown that Tyro 3 family receptors limit the magnitude and extent of macrophage activation subsequent to an initial immune stimulus. In the absence of this signaling system, macrophages are constitutively activated. This, in turn, results in a hyperactivated immune system, lymphoproliferation, the development of autoimmunity and early death.
Li J, Lin JC, Wang H, Peterson JW, Furie BC, et al. Novel Role of Vitamin K in Preventing Oxidative Injury to Developing Oligodendrocytes and Neurons. J. Neurosci. 2003;23:5816–5826.
Oxidative stress is believed to be the cause of cell death in multiple disorders of the brain, including perinatal hypoxia/ischemia. Although vitamin K is not a classical antioxidant, we report here the novel finding that vitamin K1 and K2 (menaquinone-4) potently inhibit glutathione depletion-mediated oxidative cell death in primary cultures of oligodendrocyte precursors and immature fetal cortical neurons. Neither oligodendrocytes nor neurons possess significant vitamin K-dependent carboxylase or epoxidase activity. Furthermore, the vitamin K antagonists warfarin and dicoumarol and the direct carboxylase inhibitor 2-chloro-vitamin K1 have no effect on the protective function of vitamin K against oxidative injury. Vitamin K does not prevent the depletion of intracellular glutathione caused by cystine deprivation but completely blocks free radical accumulation and cell death. The protective and potent efficacy of this naturally occurring vitamin, with no established clinical side effects, suggests a potential therapeutic application in preventing oxidative damage to undifferentiated oligodendrocytes in perinatal hypoxic/ischemic brain injury.
Liu D, Guo H, Griffin JH, Fernandez JA, Zlokovic BV. Protein S confers neuronal protection during ischemic/hypoxic injury in mice. Circulation. 2003;107:1791–6.
Pak K, Chan SL, Mattson MP (2003) Presenilin-1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. NeuroMolecular Med 3:53–64.
Pfrieger FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? BioEssays. 2003;25:72–8.
Qiu C., Karp A., von Strauss E., Winblad B., Fratiglioni L., Bellander T. Lifetime principal occupation and risk of Alzheimer's disease in the Kungsholmen project. Am J Ind Med. 2003;43:204–211.
Shankar SL, O’Guin K, Cammer M, McMorris FA, Stitt TN, Gasch RS, et al. The growth arrest-specific gene product Gas6 promotes the survival of human oligodendrocytes via a phosphatidylinositol 3-kinase dependent pathway. J Neurosci. 2003;23:4208-18.
Microarray analysis revealed that transcripts for the Axl and Mer receptor tyrosine kinases are expressed at high levels in oligodendrocytes isolated from second trimester human fetal spinal cord. In humans the sole known ligand for the Axl/Rse/Mer kinases is growth arrest-specific gene 6 (Gas6), which in the CNS is secreted by neurons and endothelial cells. We hypothesized that Gas6 is a survival factor for oligodendrocytes and receptor activation signals downstream to the phosphatidylinositol 3 (PI3)-kinase/Akt pathway to increase cell survival in the absence of cell proliferation. PI3-kinase inhibitors blocked the anti-apoptotic effect of rhGas6, whereas a MEK/ERK inhibitor had no effect. Thus Gas6 sustains human fetal oligodendrocyte viability by receptor activation and downstream signaling via the PI3-kinase/Akt pathway.
Yagami T, Ueda K, Asakura K, Okamura N, Sakaeda T, Sakaguchi G, et al. Effect of Gas6 on secretory phospholipase A2-IIA-induced apoptosis in cortical neurons. Brain Res. 2003;985:142–149.
Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, et al. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology. 2003;61(1):76-80.
Several lines of evidence suggest that inflammatory mechanisms contribute to AD. To examine whether several markers of inflammation are associated with cognitive decline in African-American and white well-functioning elders. The authors studied 3,031 African-American and white men and women (mean age 74 years) enrolled in the Health, Aging, and Body Composition Study. Serum levels of interleukin-6 (IL-6) and C-reactive protein (CRP) and plasma levels of tumor necrosis factor-alpha (TNFalpha) were measured at baseline; cognition was assessed with the Modified Mini-Mental State Examination (3MS) at baseline and at follow-up. Serum markers of inflammation, especially IL-6 and CRP, are prospectively associated with cognitive decline in well-functioning elders. These findings support the hypothesis that inflammation contributes to cognitive decline in the elderly.
Banks WA. The source of cerebral insulin. Eur J Pharmacol. 2004;490:5–12.
Björkhem I, Meaney S. Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004:806–15.
Although an immense knowledge has accumulated concerning regulation of cholesterol homeostasis in the body, this does not include the brain, where details are just emerging. Approximately 25% of the total amount of the cholesterol present in humans is localized to this organ, most of it present in myelin. Almost all brain cholesterol is a product of local synthesis, with the blood-brain barrier efficiently protecting it from exchange with lipoprotein cholesterol in the circulation. Thus, there is a highly efficient apolipoprotein-dependent recycling of cholesterol in the brain, with minimal losses to the circulation. Under steady-state conditions, most of the de novo synthesis of cholesterol in the brain appears to be balanced by excretion of the cytochrome P-450-generated oxysterol 24S-hydroxycholesterol. This oxysterol is capable of escaping the recycling mechanism and traversing the blood-brain barrier. Cholesterol levels and cholesterol turnover are affected in neurodegenerating disorders, and the capacity for cholesterol transport and recycling in the brain seems to be of importance for the development of such diseases. The possibility has been discussed that administration of inhibitors of cholesterol synthesis may reduce the prevalence of Alzheimer disease. No firm conclusions can, however, be drawn from the studies presented thus far. In the present review, the most recent advances in our understanding of cholesterol turnover in the brain are discussed.
Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–34.
CarrieÌ I. Menaquinone-4 concentration Is correlated with sphingolipid concentrations in rat brain. J Nut. 2004;134 (1):167.
Studies with animals support a role for vitamin K (VK) in the biosynthesis of sphingolipids, a class of complex lipids present in high concentrations in the brain. In mice and rats, VK deficiency decreases levels of brain sulfatides and causes behavioral alterations. In light of its heterogeneity and to better understand the role of VK in the brain, we characterized the distribution of the two main VK vitamers, phylloquinone (K1) and menaquinone-4 (MK-4), in nine distinct brain regions. The main form of VK in the brain was MK-4, and it was present in significantly higher concentrations in myelinated regions (the pons medulla and midbrain) than in nonmyelinated regions. Both regional K1 and MK-4 increased with K1 intake (P<0.05). Sphingolipid distribution varied across brain regions (P<0.001) but was not affected by K1 intake. Sphingolipids are involved in major cellular events such as cell proliferation, differentiation and survival. The strong associations reported here between brain MK-4 and sphingomyelin, sulfatides and gangliosides suggest that this vitamer may play an important role in the brain.
Chotard C, Salecker I. Neurons and glia: team players in axon guidance. Trends Neurosci. 2004;27(11):655-661.
This review focuses on studies in both Drosophila and vertebrates to highlight that mutual interactions between neurons and glia are essential in forming specific neuronal connections. Glia signal to neurons to direct pathfinding and targeting of axons, as well as to stabilize and refine axonal branches within the target area. Equally, neurons provide crucial information to glia, supporting their migration and correct positioning.
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–5.
Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481.
Alzheimer disease and type 2 diabetes are characterized by increased prevalence with aging, a genetic predisposition, and comparable pathological features in the islet and brain (amyloid derived from amyloid beta protein in the brain in Alzheimer disease and islet amyloid derived from islet amyloid polypeptide in the pancreas in type 2 diabetes). Evidence is growing to link precursors of amyloid deposition in the brain and pancreas with the pathogenesis of Alzheimer disease and type 2 diabetes, respectively. Given these similarities, we questioned whether there may be a common underlying mechanism predisposing to islet and cerebral amyloid. These data support the hypothesis that patients with Alzheimer disease are more vulnerable to type 2 diabetes and the possibility of linkage between the processes responsible for loss of brain cells and beta-cells in these diseases.
Luchsinger JA, Mayeux R. Dietary factors and Alzheimer’s disease. Lancet. 2004 Oct;3:579-857.
Alzheimer’s disease (AD) is increasing in prevalence, and environmental risk factors have not been identified with certainty. There is evidence that oxidative stress, homocysteine-related vitamins, fats, and alcohol have a role in the pathogenesis of AD. Few large epidemiological studies have explored the associations between nutrients and AD, and there has been only one trial of vitamin E in the prevention of AD. Modest to moderate alcohol intake, particularly wine, may be related to a low risk of AD. Available data do not permit definitive conclusions regarding diet and AD or specific recommendations on diet modification for the prevention of AD
Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136.
One of the most significant challenges in neuroscience is to identify the cellular and molecular processes that underlie learning and memory formation. The past decade has seen remarkable progress in understanding changes that accompany certain forms of acquisition and recall, particularly those forms which require activation of afferent pathways in the hippocampus. This progress can be attributed to a number of factors including well-characterized animal models, well-defined probes for analysis of cell signaling events and changes in gene transcription, and technology which has allowed gene knockout and overexpression in cells and animals. Of the several animal models used in identifying the changes which accompany plasticity in synaptic connections, long-term potentiation (LTP) has received most attention, and although it is not yet clear whether the changes that underlie maintenance of LTP also underlie memory consolidation, significant advances have been made in understanding cell signaling events that contribute to this form of synaptic plasticity. In this review, emphasis is focused on analysis of changes that occur after learning, especially spatial learning, and LTP and the value of assessing these changes in parallel is discussed.
Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9.
Slowly but surely, Alzheimer's disease (AD) patients lose their memory and their cognitive abilities, and even their personalities may change dramatically. These changes are due to the progressive dysfunction and death of nerve cells that are responsible for the storage and processing of information. Although drugs can temporarily improve memory, at present there are no treatments that can stop or reverse the inexorable neurodegenerative process. But rapid progress towards understanding the cellular and molecular alterations that are responsible for the neuron's demise may soon help in developing effective preventative and therapeutic strategies.
Melaragno MG, Cavet ME, Yan C, Tai LK, Jin ZG, Haendeler J & Berk BC (2004) Gas6 inhibits apoptosis in vascular smooth muscle: role of Axl kinase and Akt. J Mol Cell. Cardiol 37, 881– 887.
Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916-24.
Microglia are the resident macrophage-like population in the CNS. Microglia remain quiescent until injury or infection activates the cells to perform effector inflammatory and APC functions. Our previous studies have shown that microglia infected with a neurotropic strain of Theiler’s murine encephalomyelitis virus secreted innate immune cytokines and up-regulated costimulatory molecules and MHC class II, enabling the cells to present viral and myelin Ags to CD4 T cells. Recently, TLRs have been shown to recognize pathogen-associated molecular patterns and initiate innate immune responses upon interaction with infectious agents. We examined TLR expression on brain microglia and their functional responses upon stimulation with various TLR agonists. We report that mouse microglia express mRNA for all of the recently identified TLRs, TLR1–9, used for recognition of bacterial and viral molecular patterns. Thus, microglia appear to be a unique and important component of both the innate and adaptive immune response, providing the CNS with a means to rapidly and efficiently respond to a wide variety of pathogens. T
Seri B, Garcia-Verdugo JM, Collado-Morente L, Mcewen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J. Comp. Neurol. 2004;478;359–378.
Stenhoff J, Dahlback B & Hafizi S. Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem Biophys Res Commun. 2004;319:871– 878.
The protein product of growth arrest specific gene 6 (Gas6), is the biological ligand for the Axl subfamily of receptor tyrosine kinases. We investigated the effects of exogenous Gas6 on growth of cardiac fibroblasts isolated from genetically Gas6-deficient mice. Recombinant Gas6, containing N terminal γ-carboxyglutamic acid residues formed from a vitamin K-dependent reaction, stimulated both DNA synthesis and proliferation of cardiac fibroblasts under serum-free conditions. Gas6 also markedly enhanced survival of cells during prolonged serum starvation. Gas6 stimulated tyrosine phosphorylation of Axl as well as phosphorylation of ERK kinase. The mitogenic effects of Gas6 were inhibited by neutralising anti-Gas6 antibodies and by a soluble Axl ectodomain fusion protein. In contrast, recombinant Gas6 from cells treated with warfarin, which prevents the γ-carboxylation reaction, neither stimulated fibroblast proliferation nor activated Axl tyrosine phosphorylation. In conclusion, Gas6 appears to be a unique growth factor for fibroblasts and post-translational γ-carboxylation is necessary for its biological activity. These findings implicate vitamin K-dependent biochemical reactions in growth processes in development and in disease.
Streit WJ, Mrak RE, Griffin WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004 Jul 30;1(1):14.
Microglia make up the innate immune system of the central nervous system and are key cellular mediators of neuroinflammatory processes. Their role in central nervous system diseases, including infections, is discussed in terms of a participation in both acute and chronic neuroinflammatory responses. Specific reference is made also to their involvement in Alzheimer's disease where microglial cell activation is thought to be critically important in the neurodegenerative process.
Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63(1):8-20.
Alzheimer's disease (AD) includes etiologically heterogeneous disorders characterized by senile or presenile dementia, extracellular amyloid protein aggregations containing an insoluble amyloid precursor protein derivative, and intracytoplasmic tau protein aggregations. Recent studies also show excess neuronal aneuploidy, programmed cell death (PCD), and mitochondrial dysfunction. The leading AD molecular paradigm, the "amyloid cascade hypothesis", is based on studies of rare autosomal dominant variants and does not specify what initiates the common late-onset, sporadic form. We propose for late-onset, sporadic AD a "mitochondrial cascade hypothesis" that comprehensively reconciles seemingly disparate histopathologic and pathophysiologic features. In our model, the inherited, gene-determined make-up of an individual's electron transport chain sets basal rates of reactive oxygen species (ROS) production, which determines the pace at which acquired mitochondrial damage accumulates. Oxidative mitochondrial DNA, RNA, lipid, and protein damage amplifies ROS. In addition to defining a role for aging in AD pathogenesis, the mitochondrial cascade hypothesis also allows and accounts for overlap between the sporadic, late-onset and autosomal dominant, early onset forms of the disease.
Van Ginkel PR, Gee RL, Shearer RL, Subramanian L, Walker TM, Albert DM, et al. Expression of the receptor Tyrosine Kinase Axl promotes ocular melanoma cell survival. Cancer Res. 2004 Jan 1;64:128-134.
Metastatic tumor cells originating from cancers of a variety of tissues such as breast, skin, and prostate may remain dormant for long periods of time. In the case of uveal melanoma, the principal malignancy of the eye, complete removal of the primary tumor by enucleation can nonetheless be followed by metastatic tumor growth in distant organs months, years, or even decades later. This suggests that tumor cells have already spread to secondary sites at the time of treatment and remain dormant as micrometastases. Identifying factors that govern long-lived survival of metastatic tumor cells is therefore key to decreasing mortality associated with this and other diseases. While investigating factors in melanoma cells and normal melanocytes, we identified the receptor tyrosine kinase Axl and found up-regulation of Axl in uveal melanomas and melanoma cell lines by RNase protection, Western analysis, and immunohistochemistry. Axl has been shown to mediate cell growth and survival through its ligand Gas6 in non-transformed cells. We show that Gas6 mediates mitogenesis and cell survival in Mel 290 cells. We further demonstrate that these effects occur specifically through the Axl receptor.
Wirths O, Multaup G, Bayer TA. A modified β-amyloid hhypothesis: intraneuronal accumulation of the β-amyloid peptide- the first step of a fatal cascade. J Neurochem. 2004;91:513-520.
Accumulating evidence points to an important role of intraneuronal Aβ as a trigger of the pathological cascade of events leading to neurodegeneration and eventually to Alzheimer's disease (AD) with its typical clinical symptoms, like memory impairment and change in personality. In the present article, we review recent findings on intracellular monomeric and oligomeric β-amyloid (Aβ) generation and its pathological function in cell culture, transgenic AD mouse models and post mortem brain tissue of AD and Down syndrome patients, as well as its interaction with oxidative stress and its relevance in apoptotic cell death.
Denisova NA & Booth SL. Vitamin K and sphingolipid metabolism: evidence to date. Nutr Rev. 2005 Apr;63(4):111-21.
The brain is enriched with sphingolipids, which are important membrane constituents and major lipid signaling molecules that have a role in motor and cognitive behavior. Vitamin K has been implicated in brain sphingolipid metabolism for more than 30 years. The in vitro and in vivo studies to date suggest a role of vitamin K in the regulation of multiple enzymes involved in sphingolipid metabolism within the myelin-rich regions in the brain. However, the precise mechanisms of action are not well understood. Further, the physiological consequences of the observed effects of vitamin K on sphingolipid metabolism have not been systematically studied.
Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Neurbio. 2005;75:321-341.
Recent work in neuroscience has shown that the adult central nervous system contains neural progenitors, precursors, and stem cells that are capable of generating new neurons, astrocytes, and oligodendrocytes. While challenging previous dogma that no new neurons are born in the adult mammalian CNS, these findings bring with them future possibilities for the development of novel neural repair strategies. The purpose of this review is to present current knowledge about adult mammalian neurogenesis, to highlight the critical differences between ‘‘neurogenic’’ and ‘‘non-neurogenic’’ regions in the adult brain, and to describe the cardinal features of two well described neurogenic regions—the subventricular zone/olfactory bulb system, and the dentate gyrus of the hippocampus. The possibility of repairing neural circuitry by manipulating neurogenesis is an intriguing one, and, therefore, we also review recent efforts to understand the conditions under which neurogenesis can be induced in nonneurogenic regions of the adult CNS. This work aims toward molecular and cellular manipulation of endogenous neural precursors in situ, without transplantation.
Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129.
For approximately 80 years following Alzheimer’s description of the disease that bears his name, a gulf divided researchers who believed that extracellular deposits of the amyloid β (Aβ) peptide were pathogenic from those who believed that the deposits were secondary detritus. Since 1990, the discoveries of mutations in the Aβ peptide precursor (APP) and the APP-cleaving enzyme presenilin 1 (PS1) have enabled much progress in understanding the molecular, cellular, and tissue pathology of the aggregates that accumulate in the brains of patients with autosomal dominant familial Alzheimer disease (AD). Clarification of the molecular basis of common forms of AD has been more elusive. The central questions in common AD focus on whether cerebral and cerebrovascular Aβ accumulation is (a) a final neurotoxic pathway, common to all forms of AD; (b) a toxic by-product of an independent primary metabolic lesion that, by itself, is also neurotoxic; or (c) an inert by-product of an independent primary neurotoxic reaction.
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical Effects of ABeta Immunization (AN1792) in Patients with AD in an Interrupted Trial. Neurology. 2005;64:1553-1562.
Alzheimer’s Disease is a complex, progressive condition with symptoms that do not reveal themselves until significant changes to neuronal morphology have already occurred. The delayed manifestation of cognitive decline makes determination of the true etiological origins difficult. As a result, identification of ideal drug targets becomes seemingly impossible. The existing treatments for Alzheimer’s Disease may temporarily suppress the rate of cognitive decline, but do little to slow or halt neuronal decay. While many believe that the current approaches to identifying a cure for the disease are too narrow-minded, focusing heavily on the physical manifestations of the diseased brain such as amyloid plaques and neurofibrillary tangles, this review asserts the status of Alzheimer’s research as rational and multi-faceted.
Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201.
Hall MO, Obin MS, Heeb MJ, Burgess BL, Abrams TA. Both protein S and Gas6 stimulate outer segment phagocytosis by cultured rat retinal pigment epithelial cells. Exp Eye Res. 2005 Nov:81(5):581-591.
Survival of the retina requires the daily phagocytosis of photoreceptor outer segments (OS) by the overlying retinal pigment epithelium (RPE). OS phagocytosis by cultured RPE requires serum and we have recently shown that the vitamin K-dependent serum protein, Gas6, can completely replace serum in this process. Surprisingly, however, we show here that 4-month-old Gas6 knockout mice have normal appearing retinas, except for a reduced ratio of outer segment to inner segment length. We also show that removal of Gas6 from serum does not abrogate the ability of serum to support OS phagocytosis by rat RPE. Both of these findings suggest the presence of an additional serum ligand that is able to support OS phagocytosis by RPE cells. Protein S (PS) is a vitamin K-dependent serum protein with a high degree of structural similarity to Gas6, and a well characterized role in blood coagulation. We report here that recombinant rat PS is able to stimulate OS phagocytosis, and similar to Gas6, it does so through a Mer-dependent mechanism. This is the first demonstration of a common role for Gas6 and PS in any biological process. The existence of redundant ligands for Mer-dependent OS phagocytosis underscores the critical role of this process in the maintenance of retinal function.
Han X. Lipid alterations in the earliest clinically recognizable stage of Alzheimer's disease: Implication of the role of lipids in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 2005;2:65-77.
Lipids have many important yet distinct functions in cellular homeostasis such as forming an impermeable barrier separating intracellular and extracellular compartments, providing a matrix for the appropriate interactions of membrane-associated proteins, and serving as storage reservoirs for biologically active second messengers. Alterations in cellular lipids may therefore result in abnormal cellular functions. This review summarizes the results from the examination of lipid alterations in Alzheimers disease (AD). In addition to the effects of cholesterol on AD, substantial depletions of plasmalogen and sulfatide as well as dramatic increases in ceramide are specifically manifested at the earliest clinically recognizable stage of AD. The potential mechanism(s) underlying these changes and the potential consequences of these changes in neuronal function and in AD development are also discussed.
Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185.
The blood-brain barrier (BBB) is the regulated interface between the peripheral circulation and the central nervous system (CNS). Although originally observed by Paul Ehrlich in 1885, the nature of the BBB was debated well into the 20th century. The anatomical substrate of the BBB is the cerebral microvascular endothelium, which, together with astrocytes, pericytes, neurons, and the extracellular matrix, constitute a “neurovascular unit” that is essential for the health and function of the CNS. Disruption of BBB Tight Junctions by disease or drugs can lead to impaired BBB function and thus compromise the CNS. Therefore, understanding how BBB Tight Junctions might be affected by various factors holds significant promise for the prevention and treatment of neurological diseases.
Kang JH, Ascherio A, Grodstein F. Fruit and vegetable consumption and cognitive decline in aging women. Ann Neurol. 2005 May;57(5):713-20.
We prospectively examined fruit and vegetable intake in relation to cognitive function and decline among aging women. Participants were followed from in 1976 with biennial questionnaires, and food frequency questionnaires were administered in 1984, 1986, and every 4 years thereafter. From 1995 to 2001, we administered, by telephone, six cognitive tests measuring general cognition, verbal memory, category fluency, and working memory. Fruits were not associated with cognition or cognitive decline. However, total vegetable intake was significantly associated with less decline. Specifically, on a global score combining all tests, women in the highest quintile of cruciferous vegetables declined slower compared with the lowest quintile. Women consuming the most green leafy vegetables also experienced slower decline than women consuming the least amount (by 0.05 unit; 95% confidence interval.
Ming G. L., Song H. Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 2005;28:223–250.
Forty years since the initial discovery of neurogenesis in the postnatal rat hippocampus, investigators have now firmly established that active neurogenesis from neural progenitors continues throughout life in discrete regions of the central nervous systems (CNS) of all mammals, including humans. Significant progress has been made over the past few years in understanding the developmental process and regulation of adult neurogenesis, including proliferation, fate specification, neuronal maturation, targeting, and synaptic integration of the newborn neurons. Advances in our understanding of adult neurogenesis will not only shed light on the basic principles of adult plasticity, but also may lead to strategies for cell replacement therapy after injury or degenerative neurological diseases.
Moriya M, Nakatsuji Y, Okuno T, Hamasaki T, Sawada M, Sakoda S. Vitamin K2 ameliorates experimental autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 2005;170:11–20.
Vitamin K2 (VK2), which has been in wide use for the management of hypoprothrombinemia and osteoporosis in Japan, was tested for its efficacy on experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS). The severity of EAE was significantly ameliorated by the prophylactic administration of VK2, though it was not effective when given after the onset. Inflammatory cellular infiltration and the expression of both MHC class II and inducible nitric oxide synthase (iNOS) were reduced in the spinal cords of VK2-treated rats with EAE. The inhibitory effect of VK2 on the iNOS expression in glial cells was also observed in vitro. Considering the long use of VK2 without noticeable untoward effects, it may be applicable to the patients with MS.
Newman AB, Fitzpatrick AL, Lopez O, Jackson O, Lyketsos C, Jagust W, et al. Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study Cohort. J Am Geriatr. Soc. 2005 Jul;53(7):1101-7.
To determine whether coronary artery disease, peripheral arterial disease (PAD), or noninvasive markers of cardiovascular disease (CVD) predict the onset of dementia and Alzheimer's disease (AD). Men and women (N=3,602) with a brain magnetic resonance imaging (MRI) scan but no dementia were followed for 5.4 years. Participants with stroke were excluded. Neurologists and psychiatrists classified incident cases of dementia and subtype using neuropsychological tests, examination, medical records and informant interviews. Apolipoprotein E allele status, age, race, sex, education, Mini-Mental State Examination score, and income were assessed as potential confounders. They found that the incidence of dementia was higher in those with prevalent CVD, particularly in the subgroup with PAD. The rate of AD was 34.4 per 1,000 person-years for those with a history of CVD, versus 22.2 per 1,000 person-years without a history of CVD. Rates of AD were highest in those with PAD. Results were similar with further exclusion of those with vascular dementia from the AD group. They concluded that older adults with CVD other than stroke had a higher risk of dementia and AD than did those without CVD. The risk was highest in people with PAD, suggesting that extensive peripheral atherosclerosis is a risk factor for AD.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8.
Noel M, Reddy M. Nutrition and aging. Prim Care. 2005;32(3):659-669.
Nutritional concerns are common among older adults seen in the primary care office. The food pyramid for people over the age of 70 years is a useful starting point for discussions about what reasonably healthy older adults should be eating and drinking. If there is a decline in the ability to perform activities of daily living or if there is a decrease in appetite or the discovery of unintended weight loss, careful assessment followed by targeted interventions may improve health outcomes and the quality of life.
Sainaghi PP, Castello L, Bergamasco L, Galletti M, Bellosta P, Avanzi GC. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J Cell Physio. 2005 Jul;204(1):36-44.
Axl is a tyrosine kinase receptor and although it is expressed in malignancy such as leukemia, colon cancer, melanoma, endometrial, prostate and thyroid cancers, its role has not been completely elucidated yet and appears to be complex. The ligand of Axl, Gas6, is essential for binding. Gas6 has a mitogenic effect on several normal cell lines. We demonstrated a mitogenic activity determined by Gas6/Axl interaction in these undifferentiated metastatic human prostatic cancer cell lines.
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - Is this type 3 diabetes? J. Alzheimer’s Dis. 2005;7:63–80.
The neurodegeneration that occurs in Alzheimer’s disease (AD) is associated with a number of characteristic histopathological, molecular, and biochemical abnormalities. The inability to link these phenomena has resulted in the emergence of heavily debated theories that focus on the role of one element among others. The accumulating evidence that reduced glucose utilization and deficient energy metabolism occur early in the course of disease, suggests a role for impaired insulin signaling. The present work demonstrates extensive abnormalities in insulin and insulin-like growth factor, type I and II signaling mechanisms in brains with AD. AD may represent a neuro-endocrine disorder that resembles, yet is distinct from diabetes mellitus. Therefore, we propose the term, Type 3 Diabetes to reflect this newly identified pathogenic mechanism of neurodegeneration.
Tanz R.E. The synaptic A β hypothesis of Alzherimer’s disease. Nat. Neurosic. 2005;8:977–979.
Wu Y, Singh S, Georgescu MM & Birge RB. A role for Mer tyrosine kinase in αvβ5
integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539– 553.
Abbott NJ, Ronnback L, and Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006;7:1–53.
Batheja K, Field J. Schwann cells: origins and role in axonal maintenance and regeneration. Int J Biochem Cell Biol. 2006;38(12):1995-9.
The Schwann cell plays a vital role in maintaining the peripheral nervous system (PNS). Schwann cells are derived from neural crest cells, and come in two types either myelinating or non-myelinating Schwann cells. Both play a pivotal role in the maintenance and regeneration of axons of the neurons in the PNS. The regulation of Schwann cells is mediated a number of different neurotrophic factors which signal to transcription factors such as Krox-20, Oct-6 and Sox-10. Schwann cells are affected in a number of demyelinating disorders, such as Charcot-Marie-Tooth disease and Guillain-Barré Syndrome, infected by Mycobacterium leprae to cause leprosy and are responsible for the tumors seen in patients with neurofibromatosis type 1 and neurofibromatosis type 2. The Schwann cell is under investigation as a therapeutic agent for demyelinating diseases and spinal cord injuries. Further research on Schwann cells will help understand these diseases and perhaps lead to new treatments.
Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5 Suppl 2:61-72.
The function of mitogen-activated protein kinase (MAPK) in neurons has been the subject of considerable scrunity of late, and recent studies have given new insights into how this signaling cascade can regulate gene expression following cell-surface receptor activation. At the same time, a wealth of experimental data has demonstrated that the MAPK cascade is critically involved in the mechanisms underlying the modification of neural networks required for the stability of memories, emphasizing the high level of interest in this signaling molecule. In this review, we briefly outline the main molecular events and mechanisms of the regulation of the MAPK cascade and our understanding of the functional role of this molecular signaling cascade in regulating brain plasticity, memory consolidation and memory reconsolidation.
Freeman MR. Sculpting the nervous system: glial control of neuronal development. Curr Opin Neurobiol. 2006 Feb;16(1):119-25.
Glial cells are not passive spectators during nervous system assembly, rather they are active participants that exert significant control over neuronal development. Exciting recent studies have revealed additional ways in which glial cells also modulate neurodevelopment. Glial cells regulate the number of neurons at early developmental stages and at later stages by promoting neuronal cell death through engulfment. Glia also participate in the fine sculpting of neuronal connections by pruning excess axonal projections, shaping dendritic spines, and secreting multiple factors that promote synapse formation and functional maturation. These recent insights provide further compelling evidence that glial cells, through their diverse cellular actions, are essential contributors to the construction of a functionally mature nervous system.
Hafizi S, Dahlback B. Gas6 and Protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. The FEBS Journal. 2006;273:5231-244.
The Tyro3, Axl and Mer (TAM) receptor tyrosine kinases are activated by endogenous ligands, protein S1 (PROS1) and growth arrest-specific gene 6 (Gas6), and those have important effects on cell biology. These receptors (Rs) can be shed from the cell membrane and their soluble(s) forms can be found in plasma. We investigated the fluctuation and interactive role of TAMs and its ligands in patients with hepatocellular carcinoma (HCC), hepatitis groups, and healthy normal adult controls (NC).
Horwood JM, Dufour F, Laroche S, Davis S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006 Jun:23(12):3375-84.
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci. 2006;103:5644–51.
The premise of this review is that apolipoprotein (apo) E4 is much more than a contributing factor to neurodegeneration. ApoE has critical functions in redistributing lipids among CNS cells for normal lipid homeostasis, repairing injured neurons, maintaining synapto-dendritic connections, and scavenging toxins. In multiple pathways affecting neuropathology, including Alzheimer's disease, apoE acts directly or in concert with age, head injury, oxidative stress, ischemia, inflammation, and excess amyloid beta peptide production to cause neurological disorders, accelerating progression, altering prognosis, or lowering age of onset. We envision that unique structural features of apoE4 are responsible for apoE4-associated neuropathology.
Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS. Associations of vegetable and fruit consumption with age-related cognitive change. Neurology. 2006 Oct 24;67(8):1370-76.
This study examined the association between rates of cognitive change and dietary consumption of fruits and vegetables among older persons. The authors conducted a prospective cohort study of 3,718 participants, aged 65 years and older of the Chicago Health and Aging Project. Participants completed a food frequency questionnaire and were administered at least two of three cognitive assessments at baseline, 3-year, and 6-year follow-ups. Fruit consumption was not associated with cognitive change.cHigh vegetable but not fruit consumption may be associated with slower rate of cognitive decline with older age.
Ohsaki Y, Shirakawa H, Hiwatashi K, Furukawa Y, Mizutani T, Komai M. Vitamin K suppresses lipopolysaccharide-induced inflammation in the rat. Biosci Biotechnol Biochem. 2006;70:926–32.
In addition to the liver and bone, K is found in the brain, heart, kidney and gonadal tissue. However, the physiological role of K in these various organs is not yet fully understood. It is likely that K has functions other than its role as a cofactor of protein gamma-glutamyl carboxylation. We used in this study the DNA microarray technique to identify the effect of K status on gene expression in the rat liver. The expression of genes involved in the acute inflammation response was enhanced in rats fed with a K-deficient diet relative to the control and K1-supplemented diet groups. Moreover, dietary supplementation with K1 suppressed the inflammation induced by lipopolysaccharide administration. These results indicate that orally administrated K1 suppressed inflammation in the rat.
Peters R, Ageing and the brain. Postgrad Med J. 2006;83:84-88.
Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q, Garcia de Frutos P, et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. . 2006;33(1):96–108.
Qui C, Winblad B, Marengoni A, Klarin I, Fastbom J, Fratiglioni L. Heart failure and risk of dementia and Alzheimer disease: a population-based cohort study. Arch Intern Med. 2006 May 8;166(9):1003-8.
Heart failure has been linked to cognitive impairment in several previous studies, but to our knowledge, no investigations have explored the relationship between heart failure and the risk of dementia. We hypothesized that heart failure is a risk factor for dementia and Alzheimer disease. A community-based cohort of 1301 individuals 75 years or older and without dementia in Stockholm, Sweden, was examined 3 times over a 9-year period to detect patients with dementia and Alzheimer disease. During the 6534 person-years of follow-up (mean, 5.02 years per person), 440 subjects were diagnosed as having dementia, including 333 with Alzheimer disease. At baseline, heart failure was identified in 205 subjects. The results indicated that heart failure is associated with an increased risk of dementia and Alzheimer disease in older adults. Antihypertensive drug therapy may partially counteract the risk effect of heart failure on dementia disorders.
Shankar SL, O’Guin K, Kim M, Varnum B, Lemke G, Brosnan CF, et al. Gas6/Axl signaling activates the phosphatidylinositol 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factor alpha-induced apoptosis. J Neurosci. 2006;26:5638–48.
Sharif M, Šošic D, Rothlin CV, Kelly E, Lemke G, Olson EN, et al. Twist mediates suppression of inflammation. JEM. 2006 Aug 6;203(8):1891-1901.
Thijssen HHW, Vervoort LM, Schurgers LJ, Shearer MJ. Menadione is a metabolite of oral vitamin K. Br J Nutr. 2006 Feb;95(2):260-6.
Phylloquinone is converted into menaquinone-4 and accumulates in extrahepatic tissues. Neither the route nor the function of the conversion is known. One possible metabolic route might be the release of menadione from phylloquinone by catabolic activity. In the present study we explored the presence of menadione in urine and the effect of vitamin K intake on its excretion. Menadione in urine was analyzed by HPLC assay with fluorescence detection. Urinary menadione excretion increased greatly after oral intake of the K vitamins, phylloquinone and menaquinone-4 and -7. This effect was apparent within 1-2 h and peaked at about 3 h after intake. Amounts of menadione excreted in 24 h after vitamin K intake ranged, on a molar basis, from 1 to 5 % of the administered dose, indicating that about 5-25 % of the ingested K vitamins had been catabolized to menadione. The present study shows that menadione is a catabolic product of K vitamins formed after oral intake. The rapid appearance in urine after oral but not subcutaneous administration suggests that catabolism occurs during intestinal absorption. The observations make it likely that part of the menaquinone-4 in tissues results from uptake and prenylation of circulating menadione.
Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140.
Lipid rafts are specialized structures on the plasma membrane that have an altered lipid composition as well as links to the cytoskeleton. It has been proposed that these structures are membrane domains in which neurotransmitter signaling might occur. The effect of lipid rafts on neurotransmitter signalling has also been implicated in neurological and psychiatric diseases.
Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's disease. Exp Gerontol. 2007;42:10–21.
Craft, S. (2007). Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr. Alzheimer Res. 4, 147–152.
DelParigi A, Chen K, Salbe AD, Hill JO, Wing RR, Reiman EM, Tataranni PA. Successful dieters have increased neural activity in cortical areas involved in the control of behavior. Int J Obes (Lond). 2007 Mar;31(3):4400-8.
Elmore S. Apoptosis: a review of programmed cell death. Toxic Pathol. 2007 Jun;35(4):495-516.
The process of programmed cell death, or apoptosis, is generally characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms. Apoptosis is considered a vital component of various processes including normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, embryonic development and chemical-induced cell death. Inappropriate apoptosis (either too little or too much) is a factor in many human conditions including neurodegenerative diseases, ischemic damage, autoimmune disorders and many types of cancer. The ability to modulate the life or death of a cell is recognized for its immense therapeutic potential. Therefore, research continues to focus on the elucidation and analysis of the cell cycle machinery and signaling pathways that control cell cycle arrest and apoptosis. To that end, the field of apoptosis research has been moving forward at an alarmingly rapid rate. The goal of this review is to provide a general overview of current knowledge on the process of apoptosis including morphology, biochemistry, the role of apoptosis in health and disease, detection methods, as well as a discussion of potential alternative forms of apoptosis.
Esiri MM. Ageing and the brain. J Pathol. 2007;211:181-187.
In this review, the evidence for changes in the human brain with aging at both the macroscopic and microscopic levels is summarized. Loss of neurons is now recognized to be more modest than initial studies suggested and only affects some neuron populations. Accompanying loss of neurons is some reduction in the size of remaining neurons. This reflects a reduced size of dendritic and axonal arborizations. Some of the likely causes of these changes, including free radical damage resulting from a high rate of oxidative metabolism in neurons, glycation and dysregulation of intracellular calcium homeostasis, are discussed. The roles of genes and environmental factors in causing and responding to ageing changes are explored.
Hamilton JA, Hillard CJ, Spector AA, Watkins PA. Brain uptake and utilization of fatty acids, lipids and lipoproteins: application to neurological disorders. J Mol Neurosc. 2007;33:2-11.
Liao M.Q., Tzeng Y.J., Chang Y.X., Huang H.B., Lin T.H., Chyan C.L., Chen Y.C. The correlation between neurotoxicity, aggregative ability and secondary structure studied by sequence truncated Aβ peptides. FEBS Lett. 2007;581:1161–1165.
Aggregated beta-amyloid (Abeta) peptides are neurotoxic and cause neuronal death both in vitro and in vivo. Although the formation of a beta-sheet structure is usual required to form aggregates, the relationship between neurotoxicity and the Abeta sequence remains unclear. This article explored the correlation between Abeta sequence, secondary structure, aggregative ability, and neurotoxicity, we utilized both full-length and fragment-truncated Abeta peptides.
Matute C, Alberdi E, Domercq M, Sanchez-Gomez MV, Perez-Samartin A, Rodriguez-Antiguedad A, Perez-Cerda F (2007) Excitotoxic damage to white matter. J Anat 210:693–702.
Prieto AL, O’Dell S, Varnum B, Lai C. Localization and signaling of the receptor protein tyrosine kinase Tyro3 in cortical and hippocampal neurons. Neuroscience. 2007;150:319–34.
Tyro3 is one of three RPTKs belonging to the "TAM" receptor family, which also includes Axl and Mer. Tyro3 is the most widely expressed of these receptors in the CNS. These researchers performed a detailed study of the localization and signaling of Tyro3 polypeptides in rat hippocampal and cortical neurons. Their findings suggest that Tyro3 signaling may influence synaptic plasticity in the dendritic compartment of hippocampal and cortical neurons.
Rothlin, C. V., Ghosh, S., Zuniga, E. I., Oldstone, M. B. A., and Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034
Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109(3):1026–1033
Shatenstein B, Kergoat MJ, Reid I. Poor nutrient intakes during 1-year follow-up with community-dwelling older adults with early-stage Alzheimer dementia compared to cognitively intact matched controls. J Am Diet Assoc. 2007;107:2091-2099.
Decreased food intakes, eating behavior disturbances, and loss of body weight are particularly significant problems among those with Alzheimer dementia. This study followed the natural evolution of dietary and nutrition status among elderly community-dwelling adults with Alzheimer dementia. They found that nutrient intakes from diet and supplements were higher in control subjects, with significant differences in energy, the macronutrients, calcium, iron, zinc, vitamin K, vitamin A, and dietary fiber as well as n-3 and n-6 fatty acids. They found that dietary intakes by persons with Alzheimer dementia are poor compared to cognitively intact age-matched controls. Suboptimal diet is evident early in the onset of the disease. This vulnerable population would benefit from systematic dietary assessment and intervention to prevent further deterioration in food consumption and increased nutritional risk.
Suh H, Consiglio A, Ray J, Sawai T, D'amour KA, Gage FH. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 2007;1:515–528.
Wosik K, Cayrol R, Dodelet-Devillers A, Berthelet F, Bernard M, Moumdjian R, et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J. Neurosci. 2007; 27: 9032–9042.
Insulin is a peptide hormone involved in the regulation of glucose homeostasis. Its synthesis and function in the peripheral tissues have been extensively studied and well understood. In contrast, demonstration of insulin in the brain has raised questions concerning its origin and physiological significance. In spite of extensive studies, the source of insulin present in the brain has not yet been conclusively identified. Evidence exists in support of both peripheral and central origins of this hormone in the brain.
Zeidan YH, Hannun YA. Translational aspects of sphingolipid metabolism. Trends Mol Med. 2007;13:327–36.
Adibhatla RM, Hatcher JF. 2008. Altered lipid metabolism in brain injury and disorders. In: Quinn PJ, Wang X. Lipids in Health and Disease. Dordrecht: Springer Netherlands; 2008. p. 241–68.
Deregulated lipid metabolism may be of particular importance for CNS injuries and disorders, as this organ has the highest lipid concentration next to adipose tissue. Cholesterol is an important regulator of lipid organization and the precursor for neurosteroid biosynthesis. Low levels of neurosteroids were related to poor outcome in many brain pathologies. Apolipoprotein E is the principal cholesterol carrier protein in the brain, and the gene encoding the variant Apolipoprotein E4 is a significant risk factor for Alzheimer’s disease. The ketogenic diet where >90% of calories are derived from fat is an effective treatment for epilepsy. Understanding cytokine-induced changes in lipid metabolism will promote novel concepts and steer towards bench-to-bedside transition for therapies.
Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430-40.
Recent evidence is reviewed that glial cells are critical participants in every major aspect of brain development, function, and disease. Far more active than once thought, glial cells powerfully control synapse formation, function, and blood flow. They secrete many substances whose roles are not understood, and they are central players in CNS injury and disease. It is argued that until the roles of nonneuronal cells are more fully understood and considered, neurobiology as a whole will progress only slowly.
Bellido-Martín L, de Frutos PG. Vitamin K-dependent actions of gas-6. Vitam Horm. 2008;78:185-209.
Gas6 (growth arrestâ€specific gene 6) is the last addition to the family of plasma vitamin Kâ€dependent proteins. Gas6 was cloned and characterized in 1993 and found to be similar to the plasma anticoagulant protein S. Soon after it was recognized as a growth factorâ€like molecule, as it interacted with receptor tyrosine kinases (RTKs) of the TAM family; Tyro3, Axl, and MerTK. Since then, the role of Gas6, protein S, and the TAM receptors has been found to be important in inflammation, hemostasis, and cancer, making this system an interesting target in biomedicine. Gas6 employs a unique mechanism of action, interacting through its vitamin Kâ€dependent Gla module with phosphatidylserineâ€containing membranes and through its carboxyâ€terminal LG domains with the TAM membrane receptors. The fact that these proteins are affected by antiâ€vitamin K therapy is discussed in detail.
Binder MD, Cate HS, Prieto AL, Kemper D, Butzkueven H, Gresle MM, et al. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci. 2008;28:5195–206.
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278.
Chiu, S.-L., Chen, C.-M., and Cline, H. T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008;58: 708–719.
Conover JC, Notti RQ. The neural stem cell niche. Cell Tissue Res. 2008;331(1):211–24.
The neural stem cell niche defines a zone in which stem cells are retained after embryonic development for the production of new cells of the nervous system. This continual supply of new neurons and glia then provides the postnatal and adult brain with an added capacity for cellular plasticity, albeit one that is restricted to a few specific zones within the brain. Ultimately, based on the location of the niche, stem cells of the adult brain support regeneration in the dentate gyrus of the hippocampus and the olfactory bulb through neuron replacement.
Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 2008;8 :435-446.
Godefroy O, Jeannerod M, Allain P, Le Gall D. Frontal lobe, executive functions and cognitive control. Rev. Neurol. 2008;164:119S–S127.
Grommes C, Lee CY, Wilkinson BL, Jiang Q, Koenigsknecht-Talboo JL, Varnum B, Landreth GE. Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J Neuroimmune Pharmacol. 2008;3:130–40.
Removal of apoptotic cells is an essential process for normal development and tissue maintenance. Importantly, apoptotic cells stimulate their phagocytosis by macrophages while actively suppressing inflammatory responses. Growth arrest specific gene 6 (Gas6) is involved in this process. Animals with mutations or loss of these receptors exhibit phenotypes reflective of impaired phagocytosis and a hyperactive immune response. The present data provide direct evidence for the role of Gas6 receptors in mediating an anti-inflammatory response to ligands found on apoptotic cells with the simultaneous stimulation of phagocytosis.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A. Long-term effects of Aβ42 immunization in Alzheimer’s disease: follow-up of a randomized, placebo-controlled phase 1 trial. The Lancet. 2008;372(9634):216-223.
Immunization of patients with Alzheimer's disease with full-length amyloid-β peptide (Aβ42) can clear amyloid plaques from the brain. Our aim was to assess the relation between Aβ42 immune response, degree of plaque removal, and long-term clinical outcomes. In June, 2003, consent for long-term clinical follow-up, post-mortem neuropathological examination, or both, was sought from 80 patients (or their carers) who had entered a phase I randomized, placebo-controlled trial of immunization with Aβ42 in September, 2000. The follow-up study was completed in September, 2006. Plaques were assessed in terms of the percentage area of the cortex with Aβ immunostaining (Aβ load) and in terms of characteristic histological features reflecting plaque removal. Survival of all 80 individuals until severe dementia or death was assessed with a Cox proportional hazard model.
Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008:55(8):1265–73.
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, and O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging. 2010;31:224–243.
Nimptsch K, Rohrmann S, Linseisen J. Dietary intake of vitamin K and risk of prostate cancer in the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition (EPIC-Heidelberg). Am J Clin Nutr. 2008;87:985-992.
Okano T, Shimomura Y, Yama M, Suhara Y, Kamao M, Sugiura M. Conversion of phylloquinone (vitamin K1) into menaquinone-4 (vitamin K2) in mice. J Bio Chem. 2008;283(17):11270-79.
There are two forms of naturally occurring vitamin K, phylloquinone and the menaquinones. Phylloquinone (vitamin K(1)) is a major type (>90%) of dietary vitamin K, but its concentrations in animal tissues are remarkably low compared with those of the menaquinones, especially menaquinone-4 (vitamin K(2)), the major form (>90%) of vitamin K in tissues. Despite this great difference, the origin of tissue menaquinone-4 has yet to be exclusively defined. Our results suggest that cerebral menaquinone-4 originates from phylloquinone intake and that there are two routes of accumulation, one is the release of menadione from phylloquinone in the intestine followed by the prenylation of menadione into menaquinone-4 in tissues, and another is cleavage and prenylation within the cerebrum.
Pierce A, Bliesner B, Xu M, Nielsen-Preiss S, Lemke G, Tobet S, Wierman ME. Axl and Tyro3 modulate female reproduction by influencing gonadotropin-releasing hormone neuron survival and migration. Mol. Endocrinol. 2008;22:2481–2495.
Presse N, Shatenstein B, Kergoat MJ, Ferland G. Low vitamin K intakes in community-dwelling elders at an early stage of Alzheimer’s disease. J Am Diet Assoc. 2008;108:2095–9.
An increasing body of evidence points to a role for vitamin K in brain physiology through its participation in sphingolipid metabolism and biological activation of the vitamin K–dependent protein Gas6. One hypothesis is that vitamin K may also play a role in the pathogenesis of Alzheimer’s disease. A recent study found that patients with early-stage Alzheimer’s disease consumed less vitamin K than did cognitively intact control subjects. This study looked at the dietary intakes of vitamin K in people with early stages of AD, compared to healthy controls. They found that vitamin K intakes were significantly less in participants with Alzheimer’s disease (P0.0001), even after adjusting for energy intakes (P0.0003).
Xiong W, Chen Y, Wang H, Wang H, Wu H, Lu Q. Gas 6 and the Tyro 3 receptor tyrosine kinase subfamily regulate the phagocytic function of Sertoli cells. Reproduction. 2008;135(1):77-87.
The apoptotic spermatogenic cells and residual bodies are phagocytosed and degraded by Sertoli cells during the creation of sperm. The mechanisms of this process are largely unknown. Here, we demonstrate that Gas6 and its receptors, the Tyro 3 subfamily of receptor tyrosine kinases (RTKs; Tyro 3, Axl, and Mer), regulate the phagocytic function of Sertoli cells. The phagocytic ability of Sertoli cells increased by five times in the presence of Gas6 in serum-free medium when compared with controls. The Sertoli cells lacking Mer decrease their ability to clean up dead sperm cells. In other words, vitamin K, Gas6 and the TAMS are all involved in male fertility.
Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008 Jan 24;57(2):178-201.
The blood-brain barrier (BBB) is a highly specialized brain endothelial structure of the neurovascular system. In concert with pericytes, astrocytes, and microglia, the BBB separates components of the circulating blood from neurons. Moreover, the BBB maintains the chemical composition of the neuronal "milieu," which is required for proper functioning of neuronal circuits, synaptic transmission, synaptic remodeling, angiogenesis, and neurogenesis in the adult brain. BBB breakdown, due to disruption of the tight junctions, altered transport of molecules between blood and brain and brain and blood, aberrant angiogenesis, vessel regression, brain hypoperfusion, and inflammatory responses, may initiate and/or contribute to a "vicious circle" of the disease process, resulting in progressive synaptic and neuronal dysfunction and loss in disorders such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, multiple sclerosis, and others. This study looked at developments of new therapeutic approaches for chronic neurodegenerative disorders directed at the BBB and other nonneuronal cells of the neurovascular unit.
Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res. 2009 Apr;50 Suppl (Suppl):91-6.
Sphingolipids (SLs) are essential constituents of eukaryotic cells. Besides playing structural roles in cellular membranes, some metabolites, including ceramide, sphingosine, and sphingosine-1-phosphate, have drawn attention as bioactive signaling molecules involved in the regulation of cell growth, differentiation, senescence, and apoptosis. Understanding the many cell regulatory functions of SL metabolites requires an advanced knowledge of how and where in the cell they are generated, converted, or degraded. This review will provide a short overview of the metabolism, localization, and compartmentalization of SLs.
Binder M. D., Kilpatrick T. J. TAM receptor signalling and demyelination. Neuro-Signals. 2009;17(4):277–287.
Booth SL. Roles for vitamin K beyond coagulation. Annu. Rev. Nutr., 29 (2009), pp. 89-110.
Recent interest in vitamin K has been motivated by evidence of physiological roles beyond that of coagulation. Vitamin K and vitamin K-dependent (VKD) proteins may be involved in regulation of calcification, energy metabolism, and inflammation. However, the evidence for many of these proposed roles in the maintenance of health is equivocal. There is also an emerging viewpoint that the biochemical function of vitamin K may extend beyond that of a cofactor for the VKD carboxylation of glutamyl residues (Glus) to carboxylated Glus in VKD proteins.
Brotherson, S. Understanding Brain Development in Young Children, FS-609. n.d. NDSU Extension Service. April 2009.
Burstyn-Cohen T, Heeb MJ, Lemke G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest. 2009;119:2942–2953.
Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer's disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65.
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti, CF, Rauschka H, Schmidbauer, M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189.
Kalaria RN. Neurodegenerative disease: diabetes, microvascular pathology and Alzheimer disease. Nat. Rev. Neurol. 2009;5:305-306.
Li J, Wang H, Rosenberg PA. Vitamin K prevents oxidative cell death by inhibiting activation of 12-lipoxygenase in developing oligodendrocytes. J Neurosci Res. 2009;87:1997–2005.
Oxidative mechanisms of injury are important in many neurological disorders. Developing oligodendrocytes are particularly sensitive to oxidative stress-mediated injury. We previously demonstrated a novel function of phylloquinone (vitamin K(1)) and menaquinone 4 (MK-4; a major form of vitamin K2) in protecting developing oligodendrocytes and immature neurons against glutathione depletion-induced oxidative damage (Li et al. [ 2003] J. Neurosci. 23:5816-5826). Here we report that vitamin K at nanomolar concentrations prevents oxidative injury through blocking the activation of 12-lipoxygenase (12-LOX). In summary, our data indicate that vitamin K prevents oxidative cell death by blocking activation of 12-LOX and ROS generation.
Saller F, Brisset AC, Tchaikovski SN, Azevedo M, Chrast R, Fernandez JA, Schapira M, Hackeng TM, Griffin JH, Angelillo-Scherrer A. Generation and phenotypic analysis of protein S-deficient mice. Blood. 2009; 114:2307–14.
Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am. J. Physiol Endocrinol Metab. 2009;297:E271–E288.
2010
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. 2010;37(1):13-25.
This article reviews the blood brain barrier. Neural signalling within the central nervous system (CNS) requires a highly controlled microenvironment. Cells at three key interfaces form barriers between the blood and the CNS: the blood–brain barrier (BBB), blood–CSF barrier and the arachnoid barrier. The BBB at the level of brain microvessel endothelium is the major site of blood–CNS exchange. The structure and function of the BBB is summarized, the physical barrier formed by the endothelial tight junctions, and the transport barrier resulting from membrane transporters and vesicular mechanisms. The roles of associated cells are outlined. The embryonic development of the BBB, and changes in pathology are described. The BBB is subject to short and long-term regulation, which may be disturbed in pathology. Any programme for drug discovery or delivery, to target or avoid the CNS, needs to consider the special features of the BBB.
Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-α, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J. Leukoc. Biol. 2010;87:869–875.
To understand the role of GAS6 in modulating the immune response, we evaluated the effect on cytokine secretion by monocytes/macrophages and the molecular pathways involved. They concluded that GAS6 modulates macrophage cytokine secretion, triggering an "anti-inflammatory pathway" involving PI3K/Akt/GSK3 beta and NF-kappaB.
Bacigaluppi M, Comi G, Hermann DM. Animal models of ischemic stroke. Part two: modeling cerebral ischemia. Open Neurol J. 2010;4:34–38.
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharm. 2010;33(2):91-101.
Crivello NA, Casseus SL, Peterson JW, Smith DE, Booth SL. Age-and brain region-specific effects of dietary vitamin K on myelin sulfatides. J Nutr Biochem. 2010;11:1083-1088.
Dysregulation of myelin sulfatides is a risk factor for cognitive decline with age. Vitamin K is present in high concentrations in the brain and has been implicated in the regulation of sulfatide metabolism. This study investigated the age-related interrelation between dietary vitamin K and sulfatides in myelin fractions isolated from the brain regions of Fischer 344 male rats fed one of two dietary forms of vitamin K: vitamin K1 or its hydrogenated form, 2′,3′-dihydrophylloquinone (dK), for 28 days. Both dietary forms of vitamin K were converted to menaquinone MK4 in the brain. The efficiency of dietary dK conversion to MK-4 compared to dietary phylloquinone was lower in the striatum and cortex, and was similar to that in the hippocampus. There were significant positive correlations between sulfatides and MK-4 in the hippocampus (phylloquinone-supplemented diet, 12 and 24 months; dK-supplemented diet, 12 months) and cortex (phylloquinone-supplemented diet, 12 and 24 months). No significant correlations were observed in the striatum. Furthermore, sulfatides in the hippocampus were significantly positively correlated with MK-4 in serum. Our findings suggest that this animal model may be useful for investigation of the effect of the dietary vitamin K on sulfatide metabolism, myelin structure and behavior functions.
Božina N. The pharmacogenetics of warfarin clinical practice. Biochemia Medica. 2010;20:1.
Warfarin is the most widely prescribed oral anticoagulant. It shows great (up to 20-fold) interindividual variability in dose requirement because of both, genetic and environmental factors. Information from pharmacogenomics, a study of the interaction of the individual's genotype and drug response, can help optimize drug efficacy and minimize adverse drug reactions. Genotyping data on two genes, the warfarin metabolic enzyme CYP2C9 and warfarin target enzyme, vitamin K epoxide reductase complex 1 (VKORC1), confirmed their influence on warfarin maintenance dose. The presence of CYP2C9*2 or CYP2C9*3 variant alleles, which results in decreased enzyme activity, is associated with a significant decrease in the mean warfarin dose. VKORC1 single nucleotide polymorphisms (SNPs) explain a large fraction of the interindividual variation in warfarin dose, and VKORC1 has an approximately three-fold CYP2C9 effect. Algorithms incorporating genetic (CYP2C9 and VKORC1), demographic, and clinical factors to estimate warfarin dosage could potentially minimize the risk of overdose during warfarin induction.
Crivello NA, Casseus SL, Peterson JW, Smith DE, Booth SL. Age- and brain region-specific effects of dietary vitamin K on myelin sulfatides. J Nutr Biochem. 2010;21:1083–8.
de Chaves P, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584(9):1748-59
Simple sphingolipids such as ceramide and sphingomyelin (SM) as well as more complex glycosphingolipids play very important roles in cell function under physiological conditions and during disease development and progression. Sphingolipids are particularly abundant in the nervous system. In this review we discuss some of the functions of sphingolipids in processes that entail cellular membranes and their role in neurodegenerative diseases, with an emphasis on SM, ceramide and gangliosides.
De la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neurodegener Dis. 2010;7(1-3):116-21.
The vascular hypothesis of Alzheimer's disease (AD) which we first proposed in 1993, has become a useful concept in identifying vascular risk factors for AD or vascular dementia that can be modified through appropriate treatment to prevent, reduce or delay the onset of cognitive impairment and dementia onset. Among the more than two dozen vascular risk factors already identified for AD, are cardiovascular disease and carotid artery atherosclerosis, which may exert their pathology by chronically lowering cerebral hypoperfusion during aging. We propose and plan to initiate a clinical study to screen middle-aged, cognitively intact individuals, with carotid artery ultrasound and echocardiography to identify potentially progressive pathology in the heart and carotid artery that is considered modifiable with optimal medical treatment. This clinical strategy, if found effective in preventing pathologic conditions suspected of contributing to severe cognitive impairment, could significantly reduce AD prevalence.
Deng W., Aimone J. B., Gage F. H. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nature reviews. Neuroscience. 2010;11:339–350.
The integration of adult-born neurons into the circuitry of the adult hippocampus suggests an important role for adult hippocampal neurogenesis in learning and memory, but its specific function in these processes has remained elusive. In this article, we summarize recent progress in this area, including advances based on behavioural studies and insights provided by computational modelling. Increasingly, evidence suggests that newborn neurons might be involved in hippocampal functions that are particularly dependent on the dentate gyrus, such as pattern separation. Furthermore, newborn neurons at different maturation stages may make distinct contributions to learning and memory. In particular, computational studies suggest that, before newborn neurons are fully mature, they might function as a pattern integrator by introducing a degree of similarity to the encoding of events that occur closely in time.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010 Nov 5;3339(6005):841-5.
Microglia are the resident macrophages of the central nervous system and are associated with the pathogenesis of many neurodegenerative and brain inflammatory diseases; however, the origin of adult microglia remains controversial. In contrast to many macrophage populations, we show that microglia develop in mice that lack colony stimulating factor-1 (CSF-1) but are absent in CSF-1 receptor-deficient mice. In vivo lineage tracing studies established that adult microglia derive from primitive myeloid progenitors that arise before embryonic day 8. These results have implications for the use of embryonically derived microglial progenitors for the treatment of various brain disorders.
Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105.
The past 20 years have seen a gain in knowledge on microglia biology and microglia functions in disease that exceeds the expectations formulated when the microglia “immune network” was introduced. More than 10,000 articles have been published during this time. Important new research avenues of clinical importance have opened up such as the role of microglia in pain and in brain tumors. New controversies have also emerged such as the question of whether microglia are active or reactive players in neurodegenerative disease conditions, or whether they may be victims themselves. Premature commercial interests may be responsible for some of the confusion that currently surrounds microglia in both the Alzheimer and Parkinson’s disease research fields. Perhaps the most exciting development concerns research on the role of microglia in synaptic plasticity, which is expected to yield an answer to the question whether microglia are the brain’s electricians. This review provides an analysis of the latest developments in the microglia field.
Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathologica. 2010; 119:277–290.
Cerebral atherosclerosis (AS), small vessel disease (SVD), and cerebral amyloid angiopathy (CAA) are the most prevalent arterial disorders in the aged brain. Pathogenetically, AS and SVD share similar mechanisms. CAA, on the other hand, is characterized by the deposition of the amyloid beta-protein in the vessel wall. Despite these differences between CAA, AS and SVD, apolipoprotein E (apoE) is involved in all three disorders. Such a pathogenetic link may explain the correlations between AS, SVD, CAA, and Alzheimer's disease in the brains of elderly individuals reported in the literature. In this review, we demonstrate the relationship between AS, SVD, and CAA as well as their contribution to the development of vascular tissue lesions and we emphasize an important role for apoE in the pathogenesis of vessel disorders and vascular tissue lesions as well as for BBB dysfunction and lacunar infarct development.
Iqbal K, Liu F, Gong C-X, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010 Dec;7(8):656-664.
Tau is the major microtubule associated protein (MAP) of a mature neuron. The microtubule assembly promoting activity of tau, a phosphoprotein, is regulated by its degree of phosphorylation. Normal adult human brain tau contains 2–3 moles phosphate/mole of tau protein. Hyperphosphorylation of tau depresses this biological activity of tau. In Alzheimer disease (AD) brain tau is ∼three to four-fold more hyperphosphorylated than the normal adult brain tau and in this hyperphosphorylated state it is polymerized into neurofibrillary tangles. Some of the tau in AD brain is truncated which also promotes its self-assembly. Tau mutations found in frontotemporal dementia apparently promote its abnormal hyperphosphorylation. Inhibition of abnormal hyperphosphorylation of tau offers a promising therapeutic target for AD and related tauopathies.
Lambrecht BN, H. Hammad H. The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet. 2010;376:835-843.
Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, et al. Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci. 2010 Feb 15;123(4):595-605.
Cholesterol and sphingolipids are abundant in neuronal membranes, where they help the organization of the membrane microdomains involved in major roles such as axonal and dendritic growth, and synapse and spine stability. The aim of this study was to analyze their roles in presynaptic physiology.
Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010 Dec 15;30(50):17068-78.
The vast majority of Alzheimer's disease (AD) cases are late onset with progressive synapse loss and neurodegeneration. Although the amyloid hypothesis has generated great insights into the disease mechanism, several lines of evidence indicate that other risk factors might precondition the brain to amyloid toxicity. Here, we show that the deletion of a major lipoprotein receptor, low-density lipoprotein receptor-related protein 1 (LRP1), in forebrain neurons in mice leads to a global defect in brain lipid metabolism characterized by decreased brain levels of cholesterol, sulfatide, galactosylceramide, and triglyceride. These lipid deficits correlate with progressive, age-dependent dendritic spine degeneration, synapse loss, neuroinflammation, memory loss, and eventual neurodegeneration. We further show that the levels of glutamate receptor subunits NMDA receptor 1 and Glu receptor 1 are selectively reduced in LRP1 forebrain knock-out mice and in LRP1 knockdown neurons, which is partially rescued by restoring neuronal cholesterol. Together, these studies support a critical role for LRP1 in maintaining brain lipid homeostasis and associated synaptic and neuronal integrity, and provide important insights into the pathophysiological mechanisms in AD.
Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci Elsevier Inc.; 2010:43(1):33–42.
Matute C. Calcium dyshomeostasis in white matter pathology. Cell Calcium. 2020;47:150–157.
Calcium (Ca2+) dyshomeostasis is a major event in the pathophysiology of white matter disorders of the brain and spinal cord. All cellular components of white matter, including macroglial cells and axons, are endowed with membrane Ca2+-permeable receptors and channels lodged in the cell membrane, as well as store-operated channels and pumps. Intracellular Ca2+ overload resulting from deregulated activity of channels, such as those opened by glutamate and ATP, is deleterious to glia and axons. In this review, I summarize recent advances in our understanding of white matter Ca2+ dyshomeostasis in experimental paradigms which are relevant to stroke, perinatal ischemia, multiple sclerosis, psychiatric disorders, Alzheimer's disease and traumatic injury, and discuss some of the clinical implications of these findings.
Milhas D., Clarke C. J., Hannun Y. A. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett. 2010;584:1887–1894.
Mitew S, Kirkcaldie MTK, Halliday GM, Shepherd CE, Vickers JC, Dickson TC. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 2010 May;119(5):567-77.
We have investigated alterations in myelin associated with Amyloid beta plaques, a major pathological hallmark of Alzheimer's disease (AD), in human tissue and relevant transgenic mice models. Using quantitative morphological techniques, we determined that fibrillar amyloid beta pathology in the grey matter of the neocortex was associated with focal demyelination in human presenilin-1 familial, sporadic and preclinical AD cases, as well as in two mouse transgenic models of AD, compared with age-matched control tissue. This demyelination was most pronounced at the core of Amyloid beta plaques. Furthermore, we found a focal loss of oligodendrocytes in sporadic and preclinical AD cases associated with Abeta plaque cores. We suggest that such plaque-associated focal demyelination of the cortical grey matter might impair cortical processing.
Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, et al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010 Nov 4;468(7320):117-21.
Vitamin K occurs in the natural world in several forms, including a plant form, phylloquinone (PK), and a bacterial form, menaquinones (MKs). In many species, including humans, PK is a minor constituent of hepatic vitamin K content, with most hepatic vitamin K content comprising long-chain MKs. Menaquinone-4 (MK-4) is ubiquitously present in extrahepatic tissues, with particularly high concentrations in the brain, kidney and pancreas of humans and rats. It has consistently been shown that PK is endogenously converted to MK-4. This occurs either directly within certain tissues or by interconversion to menadione (K(3)), followed by prenylation to MK-4 (refs 9-12). Here we identify a human MK-4 biosynthetic enzyme and confirmed that the UBIAD1 gene encodes an MK-4 biosynthetic enzyme through its expression and conversion of deuterium-labelled vitamin K derivatives into MK-4. Our results show that UBIAD1 is a human MK-4 biosynthetic enzyme; this identification will permit more effective decisions to be made about vitamin K intake and bone health.
Piccinini M, Scandroglio F, Prioni S, Buccinnà B, Loberto N, Aureli M, et al. Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol Neurobiol. 2010;41:314–340.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44.
Alzheimer’s disease is the most common neurodegenerative disease encountered in patients over 60 years. By its prevalence and the consequent individual and social loss of function Alzheimer’s disease is a major public health problem. We propose a diagnostic and therapeutic discussion on this clinical problem, and should not omit the non pharmacological therapeutic approaches.
Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010 Nov 11;468(7321):253-62.
A microglial cell is both a glial cell of the central nervous system and a mononuclear phagocyte, which belongs to the haematopoietic system and is involved in inflammatory and immune responses. As such, microglia face a challenging task. The neurons of the central nervous system cannot divide and be replenished, and therefore need to be protected against pathogens, which is a key role of the immune system, but without collateral damage. In addition, after physical injury, neural cells need restorative support, which is provided by inflammatory responses. Excessive or chronic inflammatory responses can, however, be harmful. How microglia balance these demands, and how their behaviour can be modified to ameliorate disorders of the central nervous system, is becoming clear.
Reynaud E. Protein misfolding and degenerative diseases. Nature Education. 2010;3(9):28.
As our life expectancy increases, the chances of getting a degenerative disease like Alzheimer's, Parkinson's, or diabetes also increases. Why is this? As incredible as it might sound, these diseases are caused not by bacteria or viruses but rather by something conceptually quite simple: incorrect protein folding. Introductory biology courses teach us that proteins are essential for the organism because they participate in virtually every process within the cell. Therefore, if their function is impaired, the consequences can be devastating. As we age, mutations and thermodynamics (as well as some external factors) conspire against us, resulting in the misfolding of proteins. This article discusses the genetic and molecular causes for incorrect folding of proteins.
Rushworth, J. V., and Hooper, N. M. Lipid rafts: linking Alzheimer’s amyloid-β production, aggregation, and toxicity at neuronal membranes. Int. J. Alzheimers Dis. 2010 Dec 27;2011:603052.
Lipid rafts are membrane microdomains, enriched in cholesterol and sphingolipids, into which specific subsets of proteins and lipids partition, creating cell-signaling platforms that are vital for neuronal functions. Lipid rafts play at least three crucial roles in Alzheimer's Disease (AD), namely, in promoting the generation of the amyloid-β (Aβ) peptide, facilitating its aggregation upon neuronal membranes to form toxic oligomers and hosting specific neuronal receptors through which the AD-related neurotoxicity and memory impairments of the Aβ oligomers are transduced. Recent evidence suggests that Aβ oligomers may exert their deleterious effects through binding to, and causing the aberrant clustering of, lipid raft proteins including the cellular prion protein and glutamate receptors.
Sims-Robinson C, Kim B, Rosko A, and Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010;6:551–559.
Diabetes and Alzheimer disease (AD)—two age-related diseases—are both increasing in prevalence, and numerous studies have demonstrated that patients with diabetes have an increased risk of developing AD compared with healthy individuals. The underlying biological mechanisms that link the development of diabetes with AD are not fully understood. Abnormal protein processing, abnormalities in insulin signaling, dysregulated glucose metabolism, oxidative stress, the formation of advanced glycation end products, and the activation of inflammatory pathways are features common to both diseases. Hypercholesterolemia is another factor that has received attention, owing to its potential association with diabetes and AD. This review summarizes the mechanistic pathways that might link diabetes and AD.
Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol. 2010;119:7–35.
Tsiperson V, Li X, Schwartz GJ, Raine CS, Shafit-Zagardo B. GAS6 enhances repair following cuprizone-induced demyelination. PloS ONE. 2010 Dec;5(12):e15748.
Growth arrest-specific protein 6 (gas6) activities are mediated through the Tyro3, Axl, and Mer family of receptor tyrosine kinases. Gas6 is expressed and secreted by a wide variety of cell types, including cells of the central nervous system (CNS). In this study, we tested the hypothesis that administration of recombinant human Gas6 protein into the CNS improves recovery following cuprizone withdrawal. The data show that Gas6 treatment resulted in more efficient repair following cuprizone-induced injury.
Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801:806–818.
The strongest known genetic risk factor for the development of late-onset Alzheimer disease is inheritance of the apolipoprotein (apo) E4 (epsilon4 allele) although the mechanisms underlying this connection are still not entirely clear. In this review, we shall discuss the role of apo E in the brain, particularly in relation to Alzheimer disease. Cholesterol transport and homeostasis in the central nervous system (CNS) are separated from that in the peripheral circulation by the blood-brain barrier. However, the brain operates its own lipoprotein transport system that is mediated by high density lipoprotein-sized, apo E-containing lipoproteins that are synthesized and secreted by glial cells (primarily astrocytes). Apo E has also been implicated in the deposition of amyloid plaques since apo E3, more readily than apo E4, forms a complex with Ass peptides, and mediates the degradation of amyloid deposits.
Zhao X-L, Wang W-A, Tan J-X, Huang J-K, Zhang B-Z, YangCheng H-Y. Expression of β-Amyloid induced age-dependent presynaptic and axonal changes in drosophila. J Neurosci. 2010 Jan 27;30(4):1512-22.
Alzheimer's disease (AD) is attributable to synapse dysfunction and loss. This study looked at this process. They expressed wild-type or arctic form of β amyloid1-42 (Aβ) in a small group of neurons in the adult fly and performed detailed analysis of the function and structure of both axon and presynaptic terminals at the identified single-neuron level. Aβ accumulated intracellularly and induced a range of age-dependent changes, including depletion of presynaptic mitochondria, slowdown of bi-directional transports of axonal mitochondria, decreased synaptic vesicles, increased large vacuoles, and elevated synaptic fatigue. These structural and functional synaptic changes correlated with age-dependent deficit in motor behavior. All these alterations were accelerated in flies expressing the arctic form of Aβ. The depletion of presynaptic mitochondria was the earliest detected phenotype and was not caused by the change in axonal transport of mitochondria. Moreover, axonal mitochondria exhibited a dramatic reduction in number but a significant increase in size in aged Aβ-expressing flies, indicating a global depletion of mitochondria in the neuron and an impairment of mitochondria fission. These results suggest that Aβ accumulation depletes presynaptic and axonal mitochondria, leading to other presynaptic deficits.
Zhong Z, Wang Y, Guo H, Sagare A, Fernandez JA, Bell RD, et al. Protein S protects neurons from excitotoxic injury by activating the TAM receptor Tyro3- phosphatidylinositol 3-kinase-Akt pathway through its sex hormone binding globulin-like region. J Neurosci. 2010;30:15521–34.
Zhu D, Wang Y, Singh I, Bell RD, Deane R, Zhong Z, et al. Protein S controls hypoxic/ischemic blood-brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood. 2010;115:4963–4972.
Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731.
Ballard C, Gauthier S,Corbett A , Brayne C, Aarsland D, Jones E. Alzheimer's disease.Lancet. 2011;77:1019–1031.
Binder MD, Xiao J, Kemper D, Ma GZ, Murray SS, Kilpatrick TJ. Gas6 increases myelination by oligodendrocytes and its deficiency delays recovery following cuprizone-induced demyelination. PLoS ONE. 2011;6: e17727.
Bonaguidi M. A., Wheeler M. A., Shapiro J. S., Stadel R. P., Sun G. J., Ming G. L., et al. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. 2011;145:1142–1155.
Carrié I, Bélanger E, Portoukalian J, Rochford J, Ferland G. Lifelong low-phylloquinone intake is associated with cognitive impairments in old rats. J Nutr. 2011 Aug;141(8):1495-1501.
In a previous report they showed vitamin K to preferentially accumulate in brain regions rich in white matter and to positively correlate with certain sphingolipids. In this study they looked at the vitamin K status and brain function, represented by learning abilities, motor activity and anxiety in groups of rats fed a low (L=80ug/kg diet), adequate (A=500 ug/kg diet), or high (H=2000 ug/kg diet) of vitamin K1 since weaning. In 20 month old rats, sphingolipids, phylloquinone and MK4 were assessed in the cerebellum, midbrain, pons medulla, striatum and hippocampus. They found that lifetime consumption of a low-vitamin K diet results in cognitive deficits. This was associated with higher concentrations of ceramides in the hippocampus and lower gangliosides in the pons medulla and midbrain. The low vitamin K diet did not affect cognition at 6 and 12 months of age, nor did it affect motor activity or anxiety at any age. This report points to vitamin K as an important nutritional factor contributing to cognitive healthy during aging.
Cheng D, Noble J, Tang MX, Schupf N, Mayeux R, Luchsinger JA. Type 2 diabetes and late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord. 2011;31:424-430.
This study looked at type 2 diabetes (T2D) and late-onset Alzheimer’s disease (LOAD). They studied 1,488 persons aged 65 years and older without dementia at baseline from New York City. T2D was ascertained by self-report. Dementia and LOAD were ascertained by standard research procedures. The prevalence of T2D was 17%. There were 161 cases of dementia and 149 cases of LOAD. T2D was related to dementia after adjustment for age, sex, education, ethnic group and apolipoprotein E Ε4. This association was weaker when only AD – excluding cases of mixed dementia – was considered. They concluded that T2D is associated with LOAD. Cerebrovascular disease may be an important mediator.
Gao J, Cheung RT, Lee TM, Chu LW, Chan YS, Mak HK, et al. Possible retrogenesis observed with fiber tracking: an anteroposterior pattern of white matter disintegrity in normal aging and Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2011:26:47–58.
Guo H, Barrett TM, Zhong Z, Ferandez JA, Griffin JH, Freeman RS, et al. Protein S blocks the extrinsic apoptotic cascade in tissue plasminogen activator/N-methyl D-aspartate-treated neurons in Tyro3-Akt-FKHRL1 signaling pathway. Molec Neurodengeneration. 2011;6:13.
Huang X, Auinger P, Eberly S, Oakes D, Schwarzschild M, Ascherio A, Mailman R, Chen H. Serum cholesterol and the progression of Parkinson’s disease: results from DATATOP. PLoS One. 2011;6:e22854.
Lebel C, Beaulieu C. Longitudinal development of human brain wiring continues from childhood into adulthood. J Neurosci. 2011;31:10937–10947.
Lee, C.-C., Huang, C.-C., and Hsu, K.-S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011;61:867–879.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell 2011. 2010;40:619–630.
To maintain organismal homeostasis, phagocytes engulf dead cells, which are recognized as dead by virtue of a characteristic "eat me" signal exposed on their surface. The dead cells are then transferred to lysosomes, where their cellular components are degraded for reuse. Inefficient engulfment of dead cells activates the immune system, causing disease such as systemic lupus erythematosus, and if the DNA of the dead cells is not properly degraded, the innate immune response becomes activated, leading to severe anemia and chronic arthritis. Here, we discuss how the endogenous components of dead cells activate the immune system through both extracellular and intracellular pathways.
Ohsaki Y, Shirakawa H, Miura A, Giriwono PE; Sato SS, Ohashi A. et al. Vitamin K suppresses the lipopolysaccharide-induced expression of inflammatory cytokines in cultured macrophage-like cells via the inhibition of the activation of nuclear factor κB through the repression of IKKα/β phosphorylation. J. Nutr. Biochem. 2010:21:1120–1126.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011 Sep 9; 333(6048):1456-8.
Microglia are highly motile phagocytic cells that infiltrate and take up residence in the developing brain, where they are thought to provide a surveillance and scavenging function. However, although microglia have been shown to engulf and clear damaged cellular debris after brain insult, it remains less clear what role microglia play in the uninjured brain. Here, we show that microglia actively engulf synaptic material and play a major role in synaptic pruning during postnatal development in mice. These findings link microglia surveillance to synaptic maturation and suggest that deficits in microglia function may contribute to synaptic abnormalities seen in some neurodevelopmental disorders.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189.
The neuropathological hallmarks of Alzheimer disease (AD) include “positive” lesions such as amyloid plaques and cerebral amyloid angiopathy, neurofibrillary tangles, and glial responses, and “negative” lesions such as neuronal and synaptic loss. Despite their inherently cross-sectional nature, postmortem studies have enabled the staging of the progression of both amyloid and tangle pathologies, and, consequently, the development of diagnostic criteria that are now used worldwide. In addition, clinicopathological correlation studies have been crucial to generate hypotheses about the pathophysiology of the disease, by establishing that there is a continuum between “normal” aging and AD dementia, and that the amyloid plaque build-up occurs primarily before the onset of cognitive deficits, while neurofibrillary tangles, neuron loss, and particularly synaptic loss, parallel the progression of cognitive decline.
Wang J , Zhang H , Young AG , Qiu R , Argalian S , Li X , et al. Transcriptome analysis of neural progenitor cells by a genetic dual reporter strategy. Stem Cells.. 2011;29(10):1589–600.
Westhofen P., Watzka M., Marinova M., Hass M., Kirfel G., Mu J., Bevans C.G., Mu C.R., Oldenburg J. Human Vitamin K 2,3-Epoxide Reductase Complex Subunit 1-like 1 (VKORC1L1) Mediates Vitamin K-dependent Intracellular Antioxidant Function. Biol. Chem. 2011;286:15085–15094.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011 Nov 3;12(12:723-38.This article examines mechanisms of BBB dysfunction in neurodegenerative disorders, notably Alzheimer's disease, and highlights therapeutic opportunities relating to these neurovascular deficits.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012;485:512–516.
Human apolipoprotein E has three isoforms: APOE2, APOE3 and APOE4 1. APOE4 is a major genetic risk factor for Alzheimer's disease and is associated with Down's syndrome dementia and poor neurological outcome after traumatic brain injury and haemorrhage. Neurovascular dysfunction is present in normal APOE4 carriers and individuals with APOE4-associated disorders. In mice, lack of Apoe leads to blood–brain barrier (BBB) breakdown whereas APOE4 increases BBB susceptibility.
Cancela ML, ConceiÇão N, Laizé V. Gla-rich protein, a new player in tissue calcification. Adv Nutr. 2012;3:174-181.
Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nature Reviews Microbiology. 2012;10:735-42.
The intestinal microbiota consists of a vast bacterial community that resides primarily in the lower gut and lives in a symbiotic relationship with the host. A bidirectional neurohumoral communication system, known as the gut–brain axis, integrates the host gut and brain activities. Here, we describe the recent advances in our understanding of how the intestinal microbiota communicates with the brain via this axis to influence brain development and behavior. We also review how this extended communication system might influence a broad spectrum of diseases, including irritable bowel syndrome, psychiatric disorders and demyelinating conditions such as multiple sclerosis.
Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012 Nov;72(5):648-72.
The blood-brain barrier (BBB) is a term used to describe a series of properties possessed by the vasculature of the central nervous system (CNS) that tightly regulate the movement of ions, molecules, and cells between the blood and the CNS. This barrier is crucial to provide the appropriate environment to allow for proper neural function, as well as protect the CNS from injury and disease. This review discusses the cellular and molecular composition of the BBB and how the development and function of the BBB is regulated by interactions with the CNS microenvironment. They further discuss what is known about BBB dysfunction during CNS injury and disease, as well as methodology used to deliver drugs across the BBB to the CNS.
Deng T., Zhang Y., Chen Q., Yan K., Han D. Toll-like receptor-mediated inhibition of Gas6 and ProS expression facilitates inflammatory cytokine production in mouse macrophages. Immunology. 2012;135:40–50.
Ferland G. The discovery of vitamin K and its clinical applications. Ann Nutr Metab. 2012;61(3):213-8.
Vitamin K was discovered fortuitously in 1929 and was immediately associated with blood coagulation. In the decade that followed, the principal K vitamers, phylloquinone and the menaquinones, were isolated and fully characterized. In the early 1940s, the first vitamin K antagonists were discovered and crystallized with one of its derivatives, warfarin, still being widely used in today's clinical setting. However, major progress in our understanding of the mechanisms of action of vitamin K came in the 1970s with the discovery of γ-carboxyglutamic acid (Gla), a new amino acid common to all vitamin K proteins. This discovery not only provided the basis to understanding earlier findings about prothrombin but later led to the discovery of vitamin K-dependent proteins (VKDPs) not involved in hemostasis. The 1970s also saw an important breakthrough with respect to our understanding of the vitamin K cycle and marked the discovery of the first bone VKDP, osteocalcin. Important studies relating to the role of vitamin K in sphingolipid synthesis were also underway at that time and would pave the way to further work 15 years later. The decades that followed saw the discovery of additional VKDPs showing wide tissue distribution and functional scope, the latest members having been identified in 2008. The 1990s and 2000s were also marked by important epidemiological and intervention studies that focused on the translational impact of recent vitamin K discoveries, notably with respect to bone and cardiovascular health. This short review presents an overview of the history of vitamin K and of its recent developments.
Ferland G. Vitamin K and the nervous system: an overview of its actions
Adv Nutr. 2012 Mar;3(2):204-212.
The role of vitamin K in the nervous system has been somewhat neglected compared with other physiological systems despite the fact that this nutrient was identified some 40 y ago as essential for the synthesis of sphingolipids. Present in high concentrations in brain cell membranes, sphingolipids are now known to possess important cell signaling functions in addition to their structural role. In the past 20 y, additional support for vitamin K functions in the nervous system has come from the discovery and characterization of vitamin K-dependent proteins that are now known to play key roles in the central and peripheral nervous systems. Notably, protein Gas6 has been shown to be actively involved in cell survival, chemotaxis, mitogenesis, and cell growth of neurons and glial cells. Although limited in number, studies focusing on the relationship between vitamin K nutritional status and behavior and cognition have also become available, pointing to diet and certain drug treatments (i.e., warfarin derivatives) as potential modulators of the action of vitamin K in the nervous system. This review presents an overview of the research that first identified vitamin K as an important nutrient for the nervous system and summarizes recent findings that support this notion.
Gamba P, Testa G, Sottero B, Gargiulo S, Poli G, Leonarduzzi G. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann N Y Acad Sci. 2012;1259:54–64.
Gely-Pernot A, Coronas V, Harnois T, Prestoz L, Mandairon N, Didier A, et al.An endogenous vitamin K-dependent mechanism regulates cell proliferation in the brain subventricular stem cell niche. Stem Cells. 2012;30(4):719–31.
Neural stem cells (NSC) persist in the adult mammalian brain, within the subventricular zone (SVZ). The endogenous mechanisms underpinning SVZ stem and progenitor cell proliferation are not fully elucidated. Vitamin K-dependent proteins (VKDPs) are mainly secreted factors that were initially discovered as major regulators of blood coagulation. a widespread anticoagulant, is a vitamin K antagonist that inhibits the production of functional VKDP. We demonstrate that the suppression of functional VKDPs production, in vitro, by exposure of SVZ cell cultures to warfarin leads to a substantial increase in SVZ cell proliferation. We identify the anticoagulant factors, protein S and its structural homolog Gas6, as the two only VKDPs produced by SVZ cells and describe the expression and activation pattern of their Tyro3, Axl, and Mer tyrosine kinase receptors. Both in vitro and in vivo loss of function studies consisting in either Gas6 gene invalidation or in endogenous protein S neutralization, provided evidence for an important novel regulatory role of these two VKDPs in the SVZ neurogenic niche. Specifically, we show that while a loss of Gas6 leads to a reduction in the numbers of stem-like cells and in olfactory bulb neurogenesis, endogenous protein S inhibits SVZ cell proliferation. Our study opens up new perspectives for investigating further the role of vitamin K, VKDPs, and anticoagulants in NSC biology in health and disease.
Gundberg CM, Lian JB, Booth SL. Vitamin K-dependent carboxylation of osteocalcin: friend or foe? Adv Nutr. 2012;3:149-157.
Hou Y, Ouyang X, Wan R, Cheng H, Mattson MP, Cheng A. Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells. 2012 Nov;30(11):2535-47.
Kang J, Rivest S. Lipid Metabolism and Neuroinflammation in Alzheimer’s Disease: A Role for Liver X Receptors. Endocr Rev. 2012 Oct;33(5):715-46.
Liver X receptors have emerged as key regulators of lipid metabolism. In addition to their functions as cholesterol sensors, LXR have also been found to regulate inflammatory responses in macrophages. Alzheimer’s disease (AD) is a neurodegenerative disease characterized by a progressive cognitive decline associated with inflammation. Evidence indicates that the initiation and progression of AD is linked to aberrant cholesterol metabolism and inflammation. Activation of LXR can regulate neuroinflammation and decrease amyloid- peptide accumulation. Here, we highlight the role of LXR in orchestrating lipid homeostasis and neuroinflammation in the brain. In addition, diabetes mellitus is also briefly discussed as a significant risk factor for AD because of the appearing beneficial effects of LXR on glucose homeostasis. The ability of LXR to attenuate AD pathology makes them potential therapeutic targets for this neurodegenerative disease.
Kim, B., and Feldman, E. L. Insulin resistance in the nervous system. Trends Endocrino Metab. 2012;23:133–141.
Larson ME, Lesne SE. Soluble Aβ oligomer production and toxicity. J Neurochem. 2012; 120(Suppl 1):125–139.
Shearer MJ, Fu X, Booth SL. Vitamin K nutrition, metabolism, and requirements: current concepts and future research. Adv Nutr. 2012 Mar 1;3(2):182-95.
In 2001, the US Food and Nutrition Board concluded that there were insufficient data with which to establish a RDA for vitamin K. Knowledge of the relative bioavailability of multiple vitamin K forms was also poor. Since then, stable isotope methodologies have been applied to the assessment of the bioavailability of the major dietary form of vitamin K in its free state and when incorporated into a plant matrix. There is a need for stable isotope studies with enhanced sensitivity to expand knowledge of the bioavailability, absorption, disposition, and metabolism of different molecular forms of vitamin K. Another area for future research stems from evidence that common polymorphisms or haplotypes in certain key genes implicated in vitamin K metabolism might affect nutritional requirements.
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A. et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 2012;122:1316–1338.
Tarawneh R, Holtzman DM. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med. 2012;2(5).
Alzheimer disease (AD) is the most common cause of dementia in the elderly. Clinicopathological studies support the presence of a long preclinical phase of the disease, with the initial deposition of AD pathology estimated to begin approximately 10–15 years prior to the onset of clinical symptoms. The hallmark clinical phenotype of AD is a gradual and progressive decline in two or more cognitive domains, most commonly involving episodic memory and executive functions, that is sufficient to cause social or occupational impairment. Current diagnostic criteria can accurately identify AD in the majority of cases. As disease-modifying therapies are being developed, there is growing interest in the identification of individuals in the earliest symptomatic, as well as presymptomatic, stages of disease, because it is in this population that such therapies may have the greatest chance of success.
Jiang T, Yu JT, Tan L. Novel disease-modifying therapies for Alzheimer's disease. J Alzheimers Dis 2012;31:475-92.
Presently, the treatments for AD are only symptomatic and do not halt the progression of the disease. In this review, new disease-modifying therapies which reduce amyloid-β production, prevent tau hyperphosphorylation, and provide neuroprotective effects are described, including the results of in vitro and in vivo studies and clinical trials. Some typical therapies with disease-modifying effects have also been discussed.
Kalaria RN, Akinyemi R, Ihara M. Does vascular pathology contribute to Alzheimer changes? J Neurol Sci. 2012 Nov 15;322(1-2):141-7.
In recent years there has been increased interest in whether vascular disease contributes to Alzheimer's disease (AD). This review considers how modifiable risk factors such as hypertension, atherosclerosis, diabetes, dyslipidaemia and adiposity may impact on vascular structure and function to promote neurodegenerative processes and instigate AD. The presence of vascular pathology leads to chronic cerebral hypoperfusion. Pathological changes in human brain and animal studies suggest cerebral hypoperfusion which in turn induces several features of AD pathology including selective brain atrophy, white matter changes and accumulation of abnormal proteins such as amyloid β. Cerebral pathological changes may be further modified by genetic factors such as the apoliopoprotein E ε4 allele.
Laurance S, Lemarié CA, Blostein MD. Growth arrest-specific gene 6 (gas 6) and vascular hemostasis. Adv. Nutr. 2012;3:196-203.
Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489(7414):150–4.
Toledo JB, Toledo E, Weiner MW, Jack Jr CR, Jagust W, Lee VM-Y, et al. Cardiovascular risk factors, cortisol, and amyloid- β deposition in Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2012 Nov;8(6):483-489.
There is epidemiological evidence that cardiovascular risk factors (CVRF) also are risk factors for Alzheimer’s disease, but there is limited information on this from neuro-pathological studies, and even less from in vivo studies. Therefore, we examined the relationship between CVRF and amyloid-β (Aβ) brain burden Ninety-nine subjects from the Alzheimer’s Disease Neuroimaging Initiative cohort were included. They found a possible link between these CVRF and Aβ burden measured by PiB-PET. These findings highlight the utility of biomarkers to explore potential pathways linking diverse Alzheimer’s disease risk factors.
Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, et al. Vitamin K2 is a mitochondrial electron carrier that rescues Pink1 deficiency. Science. 2012 Jun 8; 336(6086):1306-10.
Human UBIAD1 localizes to mitochondria and converts vitamin K(1) to vitamin K(2). We found that vitamin K(2) was necessary and sufficient to transfer electrons in fly mitochondria. Heix mutants showed severe mitochondrial defects that were rescued by vitamin K(2), and, similar to ubiquinone, vitamin K(2) transferred electrons in fly mitochondria, resulting in more efficient adenosine triphosphate (ATP) production. Thus, mitochondrial dysfunction was rescued by vitamin K(2) that serves as a mitochondrial electron carrier, helping to maintain normal ATP production.
Winblad B et al. Safety, tolerability, and antibody response of active AB immunotherapy with CAD106 in patients with Alzheimer’s disease: randomized, double-blind, placebo-controlled, first-in-human study. Lancet Neurology. 2012 Jul;11(7):597604.
Zheng Y, Wang Q, Xiao B, Lu Q, Wang y, Wang X. Involvement of receptor tyrosine kinase Tyro3 in amyloidogenic APP processing and β-amyloid deposition in Alzheimer's disease models. PloS One. 2012;7(6):e39035.
Alquwaisani M, Buckley L, Adams C, Fanikos J. Anticoagulants: a review of the pharmacology, dosing and complications. Curr Emerg Hosp Med Rop. 2013;1(2):83-97.
Anticoagulants remain the primary strategy for the prevention and treatment of thrombosis.. Novel oral anticoagulants have emerged from clinical development and are expected to replace older agents with their ease of use and more favorable pharmacodynamic profiles. Hemorrhage is the main concerning adverse event with all anticoagulants. With their ubiquitous use, it becomes important for clinicians to have a sound understanding of anticoagulant pharmacology, dosing, and toxicity.
Carrera-Silva EA, Chan PY, Joannas L, Errasti AE, Gagliani N, Bosurgi L, et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity. 2013;39:160–170.
Dendritic cell (DC) activation is essential for the induction of immune defense against pathogens, yet needs to be tightly controlled to avoid chronic inflammation and exaggerated immune responses. T cells, once activated, produced Protein S (Pros1) that signaled through TAM receptor tyrosine kinases in DCs to limit the magnitude of DC activation. Genetic ablation of Pros1 in mouse T cells led to increased expression of costimulatory molecules and cytokines in DCs and enhanced immune responses to T cell-dependent antigens, as well as increased colitis. Additionally, PROS1 was expressed in activated human T cells, and its ability to regulate DC activation was conserved.
Chung WS , Clarke LE , Wang GX , Stafford BK , Sher A , Chakraborty C , et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400
Ferland G, Tamadon-nejad S, Ouliass B. Warfarin-induced vitamin K deficiency is associated with alterations in sphingolipid status in rats. The FASEB Journal. 2013;27(S1): DOI:10.1096/fasebj.27.1_supplement.635.12
Vitamin K (VK) is involved in sphingolipid metabolism and menaquinone-4 (MK-4), the main K vitamer in brain, is strongly correlated to cerebral sulfatides, sphingomyelin and gangliosides. Warfarin (W), a widely used oral anticoagulant, acts by blocking the VK cycle. In a previous report, we observed W-induced VK deficiency (model of Price et al. 1982) to result in cognitive deficits, an observation associated with a drastic decrease in brain MK-4. Here we report that in these rats, W-tx is associated with an alteration in sphingolipid status. Male Wistar rats were treated with warfarin for 10 wks; control rats were treated with normal water and injected with saline. Compared to the control condition, W-tx altered ceramides, sphingomyelin, sulfatides and gangliosides in specific brain regions and resulted in a loss of correlation between sphingolipids and brain MK-4. Warfarin treatment also resulted in significant changes in gangliosides GD1a and GT1b in key brain regions (p<0.05). In conclusion, W-induced VK deficiency alters sphingolipid status in brain, a finding that could contribute to the detrimental effects of W on cognition.
Gandy K., Alexa O., Canals D., Adada M., Wada M., Roddy P., et al. Sphingosine 1-phosphate induces filopodia formation through S1PR2 activation of ERM proteins. Biochem. J. 2013;449:661–672.
Giangola, M. D., Yang, W.-L., Rajayer, S. R., Nicastro, J., Coppa, G. F., Wang, P. Growth arrest-specific protein 6 attenuates neutrophil migration and acute lung injury in sepsis. Shock. 2013;40:485–491.
Han J, Tian R, Yong B, Luo C, Tan P, Shen J, et al. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem and Biophys Res Comm. 2013;435(3):493-5000.
Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, Krafft GA. The case for soluble Aβ oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol Sci. 2013 May;34(5):261-6.
Soluble Aβ oligomers are now widely recognized as key pathogenic structures in Alzheimer's disease. They inhibit synaptic function, leading to early memory deficits and synaptic degeneration, and they trigger the downstream neuronal signaling responsible for phospho-tau Alzheimer's pathology. The marginal effects observed in recent clinical studies of solanezumab, targeting monomeric Aβ, and bapineuzumab, targeting amyloid plaques, prompted expert comments that drug discovery efforts in Alzheimer's disease should focus on soluble forms of Aβ rather than fibrillar Aβ deposits found in amyloid plaques.
Hirota Y, Tsugawa N, Nakagawa K, Suhara Y, Tanaka K, Uchino Y, et al. Menadione (vitamin K3) is a catabolic product of oral phylloquinone (vitamin K1) in the intestine and a circulating precursor of tissue menaquinone-4 (vitamin K2) in rats. J Biol Chem. 2013 Nov 15;288(46):33071-80.
Mice have the ability to convert dietary phylloquinone (vitamin K1) into menaquinone-4 (vitamin K2) and store the latter in tissues. Further evidence indicates that when intravenously administered in mice phylloquinone can enter into tissues but is not converted further to menaquinone-4. These findings raise the question whether phylloquinone is absorbed and delivered to tissues in its original form and converted to menaquinone-4 or whether it is converted to menadione in the intestine followed by delivery of menadione to tissues and subsequent conversion to menaquinone-4. To answer this question, we conducted cannulation experiments using stable isotope tracer technology in rats. We confirmed that the second pathway is correct. Furthermore, high resolution of the product generated revealed that the hydroquinone, but not the quinone form of menadione, was an intermediate of the conversion. Taken together, these results provide unequivocal evidence that menadione is a catabolic product of oral phylloquinone and a major source of tissue menaquinone-4.
Hussain G, Schmitt F, Loeffler J-P, de Aguilar J-LG. Fatting the brain: a brief of recent research. Front Cell Neurosci. 2013;7:1–14.
Fatty acids are of paramount importance to all cells, since they provide energy, function as signaling molecules, and sustain structural integrity of cellular membranes. In the nervous system, where fatty acids are found in huge amounts, they participate in its development and maintenance throughout life. Growing evidence strongly indicates that fatty acids in their own right are also implicated in pathological conditions, including neurodegenerative diseases, mental disorders, stroke, and trauma. In this review, we focus on recent studies that demonstrate the relationships between fatty acids and function and dysfunction of the nervous system.
Keller RJ, Soiza K. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder. Am J Cardiovas Dis. 2013;3(4):197-226.
The etiology of Alzheimer’s disease (AD) remains unclear. The emerging view is that cerebrovascular dysfunction is a feature not only of cerebrovascular diseases, such as stroke, but also of neurodegenerative conditions, such as AD. In AD, there is impaired structure and function of cerebral blood vessels and cells in the neurovascular unit. These effects are mediated by vascular oxidative stress. Injury to the neurovascular unit alters cerebral blood flow regulation, depletes vascular reserves, disrupts the blood-brain barrier and reduces the brain’s repair capacity. Such injury can exacerbate the cognitive dysfunction exerted by incident ischemia and coexisting neurodegeneration. This article summarizes data regarding cardiovascular risk factors, vascular abnormalities and brain endothelial damage in AD.
Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013 Nov 1;5(11):a0090706.
The TAM receptors--Tyro3, Axl, and Mer--comprise a unique family of receptor tyrosine kinases, in that as a group they play no essential role in embryonic development. Instead, they function as homeostatic regulators in adult tissues and organ systems that are subject to continuous challenge and renewal throughout life. Their regulatory roles are prominent in the mature immune, reproductive, hematopoietic, vascular, and nervous systems. The TAMs and their ligands--Gas6 and Protein S--are essential for the efficient phagocytosis of apoptotic cells and membranes in these tissues; and in the immune system, they act as pleiotropic inhibitors of the innate inflammatory response to pathogens. Deficiencies in TAM signaling are thought to contribute to chronic inflammatory and autoimmune disease in humans, and aberrantly elevated TAM signaling is strongly associated with cancer progression, metastasis, and resistance to targeted therapies.
Li Q , Lu Q , Lu H , Tian S , Lu Q . Systemic Autoimmunity in TAM Triple Knockout Mice Causes Inflammatory Brain Damage and Cell Death. PLoS One. 2013;8(6).
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Aging is characterized by a progressive loss of physiological integrity, leading to impaired function and increased vulnerability to death. This deterioration is the primary risk factor for major human pathologies, including cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases. Aging research has experienced an unprecedented advance over recent years, particularly with the discovery that the rate of aging is controlled, at least to some extent, by genetic pathways and biochemical processes conserved in evolution. This Review enumerates nine tentative hallmarks that represent common denominators of aging in different organisms, with special emphasis on mammalian aging. These hallmarks are: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication.
Luchtman DW, Song C. Cognitive enhancement by omega-3 fatty acids from childhood to old age: findings from animal and clinical studies. Neuropharmacology. 2013;64:550–65.
Medhi B, Chakrabarty M. Insulin resistance: an emerging link in Alzheimer's disease. Neurol Sci. 2013;34:1719–1725.
Relentless progression of Alzheimer's disease (AD) poses a grave situation for the biomedical community to tackle. Agents starting as hot favorites in clinical trials have failed in later stages and it is time we reconsidered our approaches to intervene the disease. Quite some interesting work in the last decade has introduced a new school of thought which factors in neuronal glycemic imbalance as a major component for the development of AD. Insulin resistance in the brain has brought forward subsequent sequelae which might work towards amyloid accretion and/or tau hyperphosphorylation.
Oury F, Khrimian L, Denny CA, Gardin A, Chamouni A, Goeden N, et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell. 2013;155:228–24.
The powerful regulation of bone mass exerted by the brain suggests the existence of bone-derived signals modulating this regulation or other functions of the brain. We show here that the osteoblast-derived hormone osteocalcin crosses the blood-brain barrier, binds to neurons of the brainstem, midbrain and hippocampus, enhances the synthesis of monoamine neurotransmitters, inhibits GABA synthesis, prevents anxiety and depression and favors learning and memory independently of its metabolic functions. In addition to these post-natal functions, maternal osteocalcin crosses the placenta during pregnancy and prevents neuronal apoptosis before embryos synthesize this hormone. As a result the severity of the neuro-anatomical defects and learning and memory deficits of Osteocalcin−/− mice is determined by the maternal genotype, and delivering osteocalcin to pregnant Osteocalcin−/− mothers rescues these abnormalities in their Osteocalcin−/− progeny. This study reveals that the skeleton via osteocalcin influences cognition and contributes to the maternal influence on fetal brain development.
Presse N, Belleville S, Gaudreau P, Greenwood CE, Kergoat MJ, Morais JA, Payette H, Shatenstein B, Ferland G. Vitamin K status and cognitive function in healthy older adults. Neurobiol Aging. 2013 Dec; 34(12):2777-83.
Vitamin K modulates the synthesis and metabolism of sphingolipids, which are key players in neuronal proliferation, differentiation, senescence, cell-cell interaction, and transformation [2,3]. Recent research has linked alterations in sphingolipid metabolism to the aging process and neurodegenerative disorders such as AD. In parallel, two VKDPs, Gas6 (growth arrest-specific gene 6) and protein S, are also closely associated with the CNS functioning. Gas6 is involved in chemotaxis, mitogenesis, cell growth, and myelination, and has further been shown to rescue cortical neurons from amyloid β-induced apoptosis, a hallmark of AD. Protein S offers neuronal protection during ischemic/hypoxic injury, both in vivo and in vitro. Vitamin K may also protect neurons from N-methyl-D-aspartate-induced toxicity and apoptosis.
Popa-Wager A, Mitran S, Sivanesan S, Chang E, Buga A-M. ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev. 2013;11:963520.
The brain is a major metabolizer of oxygen and yet has relatively feeble protective antioxidant mechanisms. This paper will describe the positive aspects of moderately induced ROS but it will also outline recent research findings concerning the impact of oxidative and nitrooxidative stress on neuronal structure and function in neuropsychiatric diseases, including major depression. A common denominator of all neuropsychiatric diseases including schizophrenia and ADHD is an increased inflammatory response of the brain caused either by an exposure to proinflammatory agents during development or an accumulation of degenerated neurons, oxidized proteins, glycated products, or lipid peroxidation in the adult brain. Therefore, modulation of the prooxidant-antioxidant balance provides a therapeutic option which can be used to improve neuroprotection in response to oxidative stress. The antioxidant therapy is a novel therapeutic strategy.
Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol. Med. 2013;19:217-226.
Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013 Sep;136:2697-706.
Cerebrovascular disease and vascular risk factors are associated with Alzheimer's disease, but the evidence for their association with other neurodegenerative disorders is limited. Therefore, we compared the prevalence of cerebrovascular disease, vascular pathology and vascular risk factors in a wide range of neurodegenerative diseases and correlate them with dementia severity. Presence of cerebrovascular disease, vascular pathology and vascular risk factors was studied in 5715 cases of the National Alzheimer's Coordinating Centre database with a single neurodegenerative disease diagnosis. When cerebrovascular disease was also present, patients with Alzheimer's disease and patients with α-synucleinopathy showed relatively lower burdens of their respective lesions than those without cerebrovascular disease in the context of comparable severity of dementia at time of death. Concurrent cerebrovascular disease is a common neuropathological finding in aged subjects with dementia, is more common in Alzheimer's disease than in other neurodegenerative disorders, especially in younger subjects, and lowers the threshold for dementia which suggests that these disorders should be targeted by treatments for cerebrovascular disease.
Salian-Mehta S., Xu M., Wierman M.E. AXL and MET crosstalk to promote gonadotropin releasing hormone (GnRH) neuronal cell migration and survival. Mol. Cell. Endocrinol. 2013;374:92–100.
Tamadon-Nejad S, Ouliass B, Rochford J, Ferland G. Warfarin-induced vitamin K deficiency is associated with cognitive and behavioral perturbations, and alterations in brain sphingolipids in rats. Front Aging Neurosci. 2018 Jul;10:213.
The use of vitamin K antagonists (VKA) is associated with the onset of vascular and soft-tissue calcifications. The objective of this study was to determine whether the regular use of VKA in older adults was associated with an increased burden of intracranial calcifications compared with the use of direct oral anticoagulant (DOA). Nineteen patients aged 70 years or more using VKA for more than 3 months and 19 controls (matched for age, gender and indication for anticoagulation) using DOA for more than 3 months were consecutively included in this study. They found that the use of VKA was associated with a greater burden of intracranial calcifications compared with the use of DOA, specifically in the falx cerebri. This finding may explain part of the neurocognitive morbidity met with VKA.
Wong Ad, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The blood-brain barrier: an engineering perspective. Front Neuroeng. 2013 Aug 30;6:7.
Since Ehrlich's first experiments, only a small number of molecules, such as alcohol and caffeine have been found to cross the blood-brain barrier, and this selective permeability remains the major roadblock to treatment of many central nervous system diseases. At the same time, many central nervous system diseases are associated with disruption of the blood-brain barrier that can lead to changes in permeability, modulation of immune cell transport, and trafficking of pathogens into the brain. Over the past 10 years it has become recognized that the blood-brain barrier is a complex, dynamic system that involves biomechanical and biochemical signaling between the vascular system and the brain. Here we reconstruct the structure, function, and transport properties of the blood-brain barrier from an engineering perspective. New insight into the physics of the blood-brain barrier could ultimately lead to clinical advances in the treatment of central nervous system diseases.
Adada M., Canals D., Hannun Y. A., Obeid L. M. Sphingolipid regulation of ezrin, radixin, and moesin proteins family: implications for cell dynamics. Biochim. Biophys. Acta. 2014;1841:727–737.
Ashraf GM, Greig NH, Khan TA, Hassan I, Tabrez S, Shakil S, et al. Protein misfolding and aggregation in Alzheimer’s disease and Type 2 Diabetes Mellitus. CNS Neurol Disord Drug Targets. 2014;13(7);1280-93.
In general, proteins can only execute their various biological functions when they are appropriately folded. Their amino acid sequence encodes the relevant information required for correct three-dimensional folding, with or without the assistance of chaperones. The challenge associated with understanding protein folding is currently one of the most important aspects of the biological sciences. Misfolded protein intermediates form large polymers of unwanted aggregates and are involved in the pathogenesis of many human diseases, including Alzheimer’s disease (AD) and Type 2 diabetes mellitus (T2DM). Research data indicates that there is a widespread conformational change in the proteins involved in AD and T2DM that form β-sheets like motifs. Although conformation of these β-sheets is common to many functional proteins, the transition from α-helix to β-sheet is a typical characteristic of amyloid deposits. Any abnormality in this transition results in protein aggregation and generation of insoluble fibrils. With an abundance of common mechanisms underpinning both disorders, a significant question that can be posed is whether T2DM leads to AD in aged individuals and the associations between other protein misfolding diseases.
Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease – lessons from pathology. BMC Med. 2014 Nov 11;12:206.
Recent epidemiological and clinico-pathological data indicate considerable overlap between cerebrovascular disease (CVD) and Alzheimer's disease (AD) and suggest additive or synergistic effects of both pathologies on cognitive decline. Up to 84% of aged subjects show morphological substrates of CVD in addition to AD pathology. Further studies are warranted to elucidate the many hitherto unanswered questions regarding the overlap between CVD and AD as well as the impact of both CVD and AD pathologies on the development and progression of dementia.
Barbagallo, M., and Dominguez, L. J. Type 2 diabetes mellitus and Alzheimer's disease. World J. Diabetes. 2014;5:889–893.
Epidemiological and biological evidences support a link between type 2 diabetes mellitus (DM2) and Alzheimer's disease (AD). Persons with diabetes have a higher incidence of cognitive decline and an increased risk of developing all types of dementia. The strong epidemiological association has suggested the existence of a physiopathological link. Most recent studies have focused on the role of insulin and insulin resistance as possible links between diabetes and AD. Disturbances in brain insulin signaling mechanisms may contribute to the molecular, biochemical, and histopathological lesions in AD. Hyperglycemia itself is a risk factor for cognitive dysfunction and dementia. Hypoglycemia may also have deleterious effects on cognitive function. Recurrent symptomatic and asymptomatic hypoglycemic episodes have been suggested to cause sub-clinical brain damage, and permanent cognitive impairment. Future trials are required to clarify the mechanistic link.
Benitez A, Fieremans E, Jensen JH, Falangola MF, Tabesh A, Ferris SH, et al. White matter tract integrity metrics reflect the vulnerability of late-myelinating tracts in Alzheimer's disease. Neuro Image Clinical. 2014;4:64–71.
Biessels GJ, Strachan MWJ, Visseren FLJ, Kappelle LJ, Whitmer RA. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014 Mar;2(3):246-55.
Type 2 diabetes is associated with dementia, and also with more slight cognitive decrements. In this review we discuss trajectories from normal cognition to dementia in people with type 2 diabetes, and explore opportunities for treatment. Vascular damage is a key underlying process in both entities. Glucose-mediated processes and other metabolic disturbances might also have a role. No treatment has been established, but management of vascular risk factors and optimization of glycemic control could have therapeutic benefit.
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–43.
De Bruijn RF, Ikram MA. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Medicine. 2014;12;130.
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in elderly people, but there are still no curative options. Senile plaques and neurofibrillary tangles are considered hallmarks of AD, but cerebrovascular pathology is also common. In this review, we summarize findings on cardiovascular disease (CVD) and risk factors in the etiology of AD.
Fu SH, Zhang HF, Yang ZB, Li TB, Liu B, Lou Z, et al. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(1):87–94.
Gray SM, Meijer RI, Barrett EJ. Insulin regulates brain function, but how does it get there? Diabetes. 2014;63:3992–3997.
Hughes TM, Kuller LH, Barinas-Mitchell EJM, McDade EM, Klunk WE, Cohen AD, et al. Arterial Stiffness and β-Amyloid Progression in Nondemented Elderly Adults. 2014 May;71(5):562-568.
Recent studies show that cerebral β-amyloid (Aβ) deposition is associated with blood pressure and measures of arterial stiffness in nondemented individuals. This study examined the association between measures of arterial stiffness and change in Aβ deposition over time. The results showed that each standard deviation increase in central stiffness (carotid-femoral PWV, P = .001; heart-femoral PWV, P = .004) was linked with increases in Aβ deposition over 2 years. They concluded that Aβ deposition increases with age in nondemented individuals and that arterial stiffness is strongly associated with the progressive deposition of Aβ in the brain, especially in this age group. The association between Aβ deposition changes over time and generalized arterial stiffness indicated a relationship between the severity of subclinical vascular disease and progressive cerebral Aβ deposition.
Ji R, Meng L, Jiang X, Kumar N, Ding J, Li Q, et al. TAM receptors support neural stem cell survival, proliferation and neuronal differentiation. Plos One. 2014;9(12): e115140.
Tyro3, Axl and Mertk (TAM) receptor tyrosine kinases play multiple functional roles by either providing intrinsic trophic support for cell growth or regulating the expression of target genes that are important in the homeostatic regulation of immune responses. In the present study, we further demonstrated that all three TAM receptors were expressed by cultured primary neural stem cells (NSCs) and played a direct growth trophic role in NSCs proliferation, neuronal differentiation and survival. The cultured primary NSCs lacking TAM receptors exhibited slower growth, reduced proliferation and increased apoptosis. In addition, the neuronal differentiation and maturation of the mutant NSCs were impeded. To elucidate the underlying mechanism that the TAM receptors play on the differentiating NSCs, we examined the expression profile of neurotrophins and their receptors by real-time qPCR on the total RNAs from hippocampus and primary NSCs. The results suggest that TAM receptors support NSCs survival, proliferation and differentiation by regulating expression of neurotrophins.
Larsen JM, Martin DR, Byrne ME. Recent advances in delivery through the blood-brain barrier. Curr Top Med chem. 2014;14:1148-1160.
Current routes of delivering therapeutics to the brain to treat a variety of neurologic conditions include intracerebral, intrathecal, and intranasal delivery. Though successes have been achieved through the use of these methods, each has limitations that warrant a more universal delivery system involving the intravenous pathway. Two main barriers to intravenous delivery are the blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier. This review discusses potential methods for overcoming barriers of intravenous-mediated brain targeting as well as highlights aspects of the highly restrictive BBB anatomy that are important to consider in the design of successful drug delivery systems.
Lew ED, Oh J, Burrola PG, Lax I, Zagorska A, Traves PG, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife. 2014;3:e03385.
The TAM receptor tyrosine kinases Tyro3, Axl, and Mer regulate key features of cellular physiology, yet the differential activities of the TAM ligands Gas6 and Protein S are poorly understood. We have used biochemical and genetic analyses to delineate the rules for TAM receptor–ligand engagement. Tyro3 and Mer are activated by both ligands but only Gas6 activates Axl. Mer plays a predominant role while Axl is dispensable, and activation of Mer by Protein S is sufficient to drive phagocytosis.
Lyman M, Lloyd DF, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014 Feb;79:1-12.
Neuroinflammation is central to the common pathology of several acute and chronic brain diseases. This review examines the consequences of excessive and prolonged neuroinflammation, particularly its damaging effects on cellular and/or brain function, as well as its relevance to disease progression and possible interventions. The evidence gathered here indicates that neuroinflammation causes and accelerates long-term neurodegenerative disease, playing a central role in the very early development of chronic conditions including dementia. The wide scope and numerous complexities of neuroinflammation suggest that combinations of different preventative and therapeutic approaches may be efficacious.
Morales I, Guzman-Martinez L, Cerda-Troncoso C, Farias G, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014 Apr 22;8:112.
Alzheimer disease (AD) is the most common cause of dementia in people over 60 years old. In the search for new targets to search for novel therapeutic avenues, clinical studies in patients who used anti-inflammatory drugs indicating a lower incidence of AD have been of value to support the neuroinflammatory hypothesis of the neurodegenerative processes and the role of innate immunity in this disease. Neuroinflammation appears to occur as a consequence of a series of damage signals, including trauma, infection, oxidative agents, redox iron, oligomers of τ and β-amyloid, etc. Here, we discuss about the role of microglial and astrocytic cells, the principal agents in neuroinflammation process, in the development of neurodegenerative diseases such as AD. In this context, we also evaluated the potential relevance of natural anti-inflammatory components, which include curcumin and the novel Andean Compound, as agents for AD prevention and as a coadjuvant for AD treatments.
Mosher KR, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014 Apr 15;88(4):594-604.
Microglia, the immune cells of the central nervous system, have long been a subject of study in the Alzheimer's disease (AD) field due to their dramatic responses to the pathophysiology of the disease. With several large-scale genetic studies in the past year implicating microglial molecules in AD, the potential significance of these cells has become more prominent than ever before. As a disease that is tightly linked to aging, it is perhaps not entirely surprising that microglia of the AD brain share some phenotypes with aging microglia. In this review, we have enumerated six distinct functions of microglia and discuss the specific effects of both aging and AD.
Murai K., Qu Q., Sun G., Ye P., Li W., Asuelime G., et al. Nuclear receptor TLX stimulates hippocampal neurogenesis and enhances learning and memory in a transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9115–9120.
Nave K-A, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:5003-33.
Myelination of axons in the nervous system of vertebrates enables fast, impulse propagation, one of the best-understood concepts in neurophysiology. However, it took a long while to recognize the mechanistic complexity both of myelination by oligodendrocytes and Schwann cells and of their cellular interactions. In this review, we highlight recent advances in our understanding of myelin biogenesis, its lifelong plasticity, and the reciprocal interactions of myelinating glia with the axons they ensheath.
Pierce A.M., Keating A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014;1542:206–220.
There are 58 described RTKs, which are further categorized into 20 different RTK families. When dysregulated or overexpressed, these RTKs are implicated in disordered growth, development, and oncogenesis. The TAM family of RTKs, consisting of Tyro3, Axl, and MerTK, is prominently expressed during the development and function of the central nervous system (CNS). Aberrant expression and dysregulated activation of TAM family members has been demonstrated in a variety of CNS-related disorders and diseases.
Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014 Mar;55(3):345-362.
In contrast to other fat-soluble vitamins, dietary vitamin K is rapidly lost to the body resulting in comparatively low tissue stores. Deficiency is kept at bay by the ubiquity of vitamin K in the diet, synthesis by gut microflora in some species, and relatively low vitamin K cofactor requirements for γ-glutamyl carboxylation. However, as shown by fatal neonatal bleeding in mice that lack vitamin K epoxide reductase (VKOR), the low requirements are dependent on the ability of animals to regenerate vitamin K from its epoxide metabolite via the vitamin K cycle. The identification of the genes encoding VKOR and its paralog VKOR-like 1 (VKORL1) has accelerated understanding of the enzymology of this salvage pathway. In parallel, a novel human enzyme that participates in the cellular conversion of phylloquinone to menaquinone (MK)-4 was identified as (UBIAD1). Recent studies suggest that side-chain cleavage of oral phylloquinone occurs in the intestine, and that menadione is a circulating precursor of tissue MK-4. The mechanisms and functions of vitamin K recycling and MK-4 synthesis have dominated advances made in vitamin K biochemistry over the last five years and, after a brief overview of general metabolism, are the main focuses of this review.
Tsou W-I, Nguyen K-QN, Calarese DA, Garforth SJ, Antes AL, Smirnov S V, et al. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-induced Activation. J Biol Chem. 2014;289(37):25750–63.
Van der Meer JHM, Van der Poll T, Van’t Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123(16):2460-2469.
TAM receptors (Tyro3, Axl, and Mer) belong to a family of receptor tyrosine kinases that have important effects on hemostasis and inflammation. Also, they affect cell proliferation, survival, adhesion, and migration. TAM receptors can be activated by the vitamin K–dependent proteins Gas6 and protein S. When activated, the TAM receptors have effects on primary hemostasis and coagulation and display an anti-inflammatory or a proinflammatory effect, depending on cell type. Because of the effects TAM receptors have on hemostasis, inflammation, and cancer growth, their modulation could make interesting therapeutic targets in thromboembolic disease, atherosclerosis, sepsis, autoimmune disease, and cancer.
Weimar C, Winkler A, Dlugaj M, Lehmann N, Hennig F, Bauer M, et al. Ankle-Briachial index but neither intima media thickness nor coronary artery calcification is associated with mild cognitive impairment. J Alzheimers Dis. 2014;47(2):433-42.
Several studies have reported an association of atherosclerosis with mild cognitive impairment (MCI) and dementia independent of cardiovascular risk factors. This study looked at artery thickness and calcification and found that the degree of generalized atherosclerosis as measured by the ABI is associated with MCI and with naMCI in a population-based cohort.
Wood WG, Li L, Müller WE, Eckert GP. Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem. 2014:559–72.
Yun J, Finkel. Mitohormesis. Cell Metab. 2014 May 6;19(5):757-66.
For many years, mitochondria were viewed as semiautonomous organelles, required only for cellular energetics. This view has been largely supplanted by the concept that mitochondria are fully integrated into the cell and that mitochondrial stresses rapidly activate cytosolic signaling pathways that ultimately alter nuclear gene expression. Remarkably, this coordinated response to mild mitochondrial stress appears to leave the cell less susceptible to subsequent perturbations. This response, termed mitohormesis, is being rapidly dissected in many model organisms. A fuller understanding of mitohormesis promises to provide insight into our susceptibility for disease and potentially provide a unifying hypothesis for why we age.
Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol (2014) 15(10):920–8.
Zuo P-Y, Chen X-L, Lei Y-H, Liu C-Y, Liu Y-W. Growth arrest-specific gene 6 protein promotes the proliferation and migration of endothelial progenitor cells through the PI3K/AKT signaling pathway. Int J Mol Med. 2014;34:299–306.
Annweiler, C.; Denis, S.; Duval, G.; Ferland, G.; Bartha, R.; Beauchet, O. Use of vitamin K antagonists and brain volumetry in seniors: Preliminary results from the GAIT study. J. Am Ger Soc. 2015;10(63):10.
Older adults use vitamin K antagonists (VKAs) widely for the prophylaxis and treatment of thromboembolic diseases. VKAs reduce the bioavailability of the active form of vitamin K, but recent data suggest that vitamin K could positively influence brain function, particularly by regulating the synthesis of sphingolipids, which are constituents of the myelin sheath and neuronal membrane, and by regulating the biological activation of vitamin K–dependent proteins (VKDPs), which are involved in neuronal health and function. Epidemiological data indicate a positive association between serum vitamin K concentrations and episodic memory function in older adult. In contrast, use of VKAs was associated with cognitive impairment in older adults, regardless of atrial fibrillation or stroke. The objective of this study was to determine whether the use of VKAs was associated with lower brain volume in older adult. They found that the use of VKA by older adults was associated with smaller brain volume normalized to intracranial volume; specifically, lower volumes of gray matter were found, including in the temporal cortex and hippocampus.
Annweiler C, Ferland G, Barberger-Gateau P, Brangier A, Rolland Y, Beauchet O. Vitamin K antagonists and cognitive impairment: Results from a cross-sectional pilot study among geriatric patients. J Gerontol A Biol Sci Med Sci. 2015a;70:97–101.
This study looked to determine whether using vitamin K antagonists (VKAs) was associated with cognitive impairment among geriatric patients. They found that the risk of cognitive impairment was 15% higher with VKAs, specifically with fluindione. They found more frequent cognitive impairment associated with the use of VKAs, specifically fluindione, among geriatric patients.
Aureli M, Grassi S, Prioni S, Sonnino S, Prinetti A. Lipid membrane domains in the brain. Biochimica et Biophysica Acta Molecular and Cell Biology of Lipids. 2015 Aug;1851(8):1006-1016.
Lipid-driven membrane domains function as dynamic platforms for signal transduction, protein processing, and membrane turnover. Essential events involved in the development and in the maintenance of the functional integrity of the brain depend on the organization of lipid-driven membrane domains, and alterations in lipid homeostasis, leading to deranged lipid-driven membrane organization, are common in several major brain diseases. In this review, we summarize the forces behind the formation of lipid membrane domains and their biological roles in different brain cells.
Bos D, Vernooij MW, de Bruijn RFAG, Koudstaal PJ, Hofman A, Franco OH, van der Lugt A, et al. Atherosclerotic calcification is related to a higher risk of dementia and cognitive decline. Alzheimers Dement. 2015 Jun:11(6):639-47.
Between 2003-2006, 2364 nondemented persons underwent computed tomography of the coronaries, aortic arch, extracranial, and intracranial carotid arteries to quantify atherosclerotic calcification. Participants were followed for incident dementia (n = 90) until April 2012. At baseline and follow-up participants also underwent a cognitive test battery. They found that atherosclerosis, in particular in the extracranial carotid arteries, is related to a higher risk of dementia and cognitive decline.
Braniste V., Al-Asmakh M., Kowal C., Anuar F., Abbaspour A., Tóth M., Korecka A., Bakocevic N., Ng L.G., Kundu P., et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014;6:263ra158.
Cermenati G, Mitro N, Audano M, Melcangi RC, Crestani M, De Fabiani E, et al. Lipids in the nervous system: from biochemistry and molecular biology to patho-physiology. Biochim Biophys Acta - Mol Cell Biol Lipids 2015;1851:51–60.
Lipids in the nervous system accomplish a great number of key functions, from synaptogenesis to impulse conduction, and more. Most of the lipids of the nervous system are localized in myelin sheaths. It has long been known that myelin structure and brain homeostasis rely on specific lipid-protein interactions and on specific cell-to-cell signaling. Key findings recently emerged in these areas are here summarized. In addition, they briefly discuss how this new knowledge can open novel approaches for the treatment of diseases associated with alteration of lipid metabolism/homeostasis in the nervous system.
Chamouni A, Schreiweis C, Oury F. Bone, brain & beyond. Rev Endocr Metab Disord. 2015;6:99-113.
Chouet J, Ferland G, Féart C, Rolland Y, Presse N, Boucher K, et al. Dietary vitamin K intake is associated with cognition and behaviour among geriatric patients: The CLIP study. Nutrients. 2015;7:6739-6750.
Our objective was to determine whether dietary vitamin K intake was associated with cognition and behavior among older adults. 192 consecutive participants over 65 years, recruited in the cross-sectional CLIP (Cognition and LIPophilic vitamins) study, were separated into two groups according to the tertiles of dietary phylloquinone intake (i.e., lowest third below 207 µg/day versus the other two thirds combined). Compared to participants in the lowest third of dietary phylloquinone intake, those with higher intake had higher (i.e., better) mean MMSE score and lower (i.e., better) FBRS score. They found that higher dietary phylloquinone intake was associated with better cognition and behavior among older adults.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015 Jan;7(1):a020412.
Blood vessels are critical to deliver oxygen and nutrients to all of the tissues and organs throughout the body. The blood vessels that vascularize the central nervous system (CNS) possess unique properties, termed the blood-brain barrier, which allow these vessels to tightly regulate the movement of ions, molecules, and cells between the blood and the brain. This precise control of CNS homeostasis allows for proper neuronal function and also protects the neural tissue from toxins and pathogens, and alterations of these barrier properties are an important component of pathology and progression of different neurological diseases. The physiological barrier is coordinated by a series of physical, transport, and metabolic properties possessed by the endothelial cells (ECs) that form the walls of the blood vessels, and these properties are regulated by interactions with different vascular, immune, and neural cells. Understanding how these different cell populations interact to regulate the barrier properties is essential for understanding how the brain functions during health and disease.
DiNicolantonio JJ, Bhutani J, O’Keefe JH. The health benefits of vitamin K. Open Heart. 2015;2:30003000.
Vitamin K has important functions within the body, some of which are still being discovered. Research has shown that vitamin K is an anticalcification, anticancer, bone-forming and insulin-sensitising molecule. Recent data indicate that subclinical vitamin K deficiency is not uncommon. Additionally, vitamin K antagonists such as warfarin may cause detrimental side effects, which may partly be blunted through vitamin K supplementation.
Ecker C, Bookheimer SY, Murphy DGM. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015:1121–34.
Erny D., HrabÄ› de Angelis A.L., Jaitin D., Wieghofer P., Staszewski O., David E., Keren-Shaul H., Mahlakoiv T., Jakobshagen K., Buch T., et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015;18:965–977.
Franklin RJM, Goldman SA. Glia disease and repair-remyelination. Cold Spring Harbor 451 perspectives in biology. 2015;7(7):a020594.
The inability of the mammalian central nervous system (CNS) to undergo spontaneous regeneration has long been regarded as a central tenet of neurobiology. However, although this is largely true of the neuronal elements of the adult mammalian CNS, save for discrete populations of granular neurons, the same is not true of its glial elements. In particular, the loss of oligodendrocytes, which results in demyelination, triggers a spontaneous and often highly efficient regenerative response, remyelination, in which new oligodendrocytes are generated and myelin sheaths are restored to denuded axons. In this review, we will review the biology of remyelination, including the cells and signals involved; describe when remyelination occurs and when and why it fails and the consequences of its failure; and discuss approaches for therapeutically enhancing remyelination in demyelinating diseases of both children and adults.
Fujii S, Shimizu A, Takeda N, Oguchi K, Katsurai T. Systematic synthesis and anti-inflammatory activity of x-carboxylated menaquinone derivatives. Investigations on identified and putative vitamin K2 metabolites. Bioorg. Med. Chem. 2015;23:2344–2352.
Ginisity A, Gély-Pernot A, , Abaamrane L, Morel F, Arnault P, Coronas V. Evidence for a subventricular zone neural stem cell phagocytic activity stimulated by the vitamin K-dependent factor Protein S. Stem Cells 2015;33:515–525.
Neural stem cells, whose major reservoir in the adult mammalian brain is the subventricular zone (SVZ), ensure neuropoiesis, a process during which many generated cells die. Removal of dead cells and debris by phagocytes is necessary for tissue homeostasis. We show that SVZ cell phagocytic activity is an active process, and is stimulated by the vitamin K-dependent factor protein S (ProS). ProS neutralizing antibodies inhibit SVZ cell phagocytic activity and exposure of SVZ cells to apoptotic cell-derived fragments induces a transient Mer tyrosine kinase receptor (MerTK) phosphorylation. By revealing that neural stem-like cells act within the SVZ neurogenic niche as phagocytes and that the ProS/MerTK path represents an endogenous regulatory mechanism for SVZ cell phagocytic activity, the present report opens-up new perspectives for both stem cell biology and brain physiopathology.
Huang, C. C., Chung, C. M., Leu, H. B., Lin, L. Y., Chiu, C. C., Hsu, C. Y., et al. Diabetes mellitus and the risk of Alzheimer's disease: a nationwide population-based study. PLoS ONE. 2014;9:e87095.
Jembrek MJ, Hot PF, Šimić G. Ceramides in Alzheimer’s disease:Key mediators of neuronal apoptosis induced by oxidative stress and Aβ Accumulation. Oxidative Med and Cell Longevity. 2015;2015:346783.
Alzheimer’s disease (AD), the most common chronic and progressive neurodegenerative disorder, is characterized by extracellular deposits of amyloid β-peptides (Aβ) and intracellular deposits of hyperphosphorylated tau (phospho-tau) protein. Ceramides, the major molecules of sphingolipid metabolism and lipid second messengers, have been associated with AD progression and pathology via Aβ generation. Ceramides may initiate a cascade of biochemical alterations, which ultimately leads to neuronal death. This review summarizes recent findings related to the role of ceramides in oxidative stress-driven neuronal apoptosis and interplay with Aβ in the cascade of events ending in neuronal degeneration.
Leblhuber F, Geisler S, Steiner K, Fuchs D, Schütz B. Elevated fecal calprotectin in patients with Alzheimer’s dementia indicates leaky gut. J Neural Transm. 2015;122:1319–1322.
Li L, Tao Y, Tang J, Chen Y, Yang Z, Feng Y, et al. A cannabinoid receptor 2 agonist prevents thrombin-induced blood-brain barrier damage via the inhibition of microglial activation and matrix metalloproteinase expression in rats. Transl Stroke Res. 2015;6:467-77.
Li X, Song D, Leng SX. Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment. Clin Interv Aging. 2015 Mar 10;10_549-60.
The aim of this paper is to provide a comprehensive review of the epidemiological evidence linking type 2 diabetes mellitus and its related conditions, including obesity, hyperinsulinemia, and metabolic syndrome, to Alzheimer's disease (AD). Several mechanisms could help to explain this proposed link; however, our focus is on insulin resistance and deficiency. Studies have shown that insulin resistance and deficiency can interact with amyloid-β protein and tau protein phosphorylation, each leading to the onset and development of AD. Based on those epidemiological data and basic research, it was recently proposed that AD can be considered as "type 3 diabetes". It is worth mentioning that the therapeutic effects of treatment rugs are influenced by the apolipoprotein E (APOE)-ε4 genotype. Patients without the APOE-ε4 allele showed better treatment effects than those with this allele.
Magnusson KR, Hauck L, Jeffrey BM, Elias V, Humphrey A, Nath R, et al. Relationships between diet-related changes in the gut microbiome and cognitive flexibility. Neuroscience. 2015 Aug 6;300:128-40.
Western diets are high in fat and sucrose and can influence behavior and gut microbiota. There is growing evidence that altering the microbiome can influence the brain and behavior. This study was designed to determine whether diet-induced changes in the gut microbiota could contribute to alterations in anxiety, memory or cognitive flexibility. Some similarities in alterations in the microbiome were seen in both the high-fat and high-sucrose diets (e.g., increased Clostridiales), as compared to the normal diet, but the percentage decreases in Bacteroidales were greater in the high-sucrose diet mice. Lactobacillales was only significantly increased in the high-sucrose diet group and Erysipelotrichales was only significantly affected by the high-fat diet. The high-sucrose diet group was significantly impaired in early development of a spatial bias for long-term memory, short-term memory and reversal training, compared to mice on normal diet. There was no significant effect of diet on step-down, exploration or novel recognitions. Higher percentages of Clostridiales and lower expression of Bacteroidales in high-energy diets were related to the poorer cognitive flexibility in the reversal trials. These results suggest that changes in the microbiome may contribute to cognitive changes associated with eating a Western diet.
May CD, Garnett J, Ma XY, Landers SM, Ingram DR, Demicco EG, et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer. 2015;15:901.
Nhan HS, Chiang K, Koo EH. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol. 2015:129(1):1–19.
Pasquier F, Sadowsky C, Holstein A, LePrince Leterme G, Peng Y, Jackson N, et al. Two Phase w multiple ascending-dose studies of Vanutide Cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J Alzheimers Dis. 2015;51(4):1131-43.
Vanutide cridificar (ACC-001), an immunotherapeutic vaccine, is a potentially disease-modifying therapy that aims to reduce brain amyloid-β (Aβ) plaques in patients with Alzheimer's disease (AD). ACC-001 was evaluated in two phase 2a, multicenter, randomized, third party-unblinded, placebo-controlled, multiple ascending-dose studies of ACC-001 (3μg, 10μg, 30μg) with and without QS-21 adjuvant that enrolled patients with mild-to-moderate AD (n = 245). Exploratory cognitive evaluations, volumetric brain MRI, and CSF biomarkers did not show differences or trends between treatment groups and placebo. ACC-001 with or without QS-21 adjuvant has an acceptable safety profile in patients with mild-to-moderate AD.
Paul R, Choudhury A, Borah A, Cholesterol - A. Putative endogenous contributor towards Parkinson’s disease. Neurochem. Int Elsevier Ltd. 2015:125–33.
Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, et al. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci. 2015 Jul 8; 35(27):9977-89.
With severe injury or disease, microglia become chronically activated and damage the local brain environment, likely contributing to cognitive decline. We previously discovered that microglia are dependent on colony-stimulating factor 1 receptor (CSF1R) signaling for survival in the healthy adult brain, and we have exploited this dependence to determine whether such activated microglia contribute deleteriously to functional recovery following a neuronal lesion. Here, we induced a hippocampal lesion in mice producing both a neuroinflammatory reaction and behavioral alterations. Collectively, we demonstrate that microglia exert beneficial effects during a diphtheria toxin-induced neuronal lesion, but impede recovery following insult.
Rossor M, Collinge J, Fox N, Mead S, Mummery C, Rohrer J, et al. 2016. Dementia and cognitive impairment. In Clarke C, Howard R, Rossor M, Shorvon S, editors. Neurology: A Queen Square Textbook. 2nd ed. Volume 30. Hoboken, NJ, USA: John Wiley & Sons, Ltd. p. 289–336.
Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355-91.
The TAM receptor tyrosine kinases (RTKs)-TYRO3, AXL, and MERTK-together with their cognate agonists GAS6 and PROS1 play an essential role in the resolution of inflammation. Deficiencies in TAM signaling have been associated with chronic inflammatory and autoimmune diseases. Three processes regulated by TAM signaling may contribute, either independently or collectively, to immune homeostasis: the negative regulation of the innate immune response, the phagocytosis of apoptotic cells, and the restoration of vascular integrity. Recent studies have also revealed the function of TAMs in infectious diseases and cancer. Here, we review the important milestones in the discovery of these RTKs and their ligands and the studies that underscore the functional importance of this signaling pathway in physiological immune settings and disease.
Spagnuolo MS, Mollica MP, Maresca B, Cavaliere G, Cefaliello C, Trinchese G, et al. High fat diet and inflammation – modulation of haptoglobin level in rat brain. Front Cell Neurosci. 2015;9:479.
Tang Y, Wu S, Liu Q, Xie J, Zhang J, Han D, et al. Mertk deficiency affects macrophage directional migration via disruption of cytoskeletal organization. PLoS ONE. 2015:10:e0117787.
Viola K.L., Klein W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129:183–206.
Protein aggregation is common to dozens of diseases including prionoses, diabetes, Parkinson's and Alzheimer's. Over the past 15 years, there has been a paradigm shift in understanding the structural basis for these proteinopathies. Precedent for this shift has come from investigation of soluble Aβ oligomers (AβOs), toxins now widely regarded as instigating neuron damage leading to Alzheimer's dementia. Overall, current evidence indicates that Aβ oligomers provide a substantive molecular basis for the cause, treatment and diagnosis of Alzheimer's disease.
Wang W-Y, Tan M-S, Yu J-T, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015 Jun;3(10);136.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder of the brain, which is characterized by the formation of extracellular amyloid plaques (or senile plaques) and intracellular neurofibrillary tangles. However, increasing evidences demonstrated that neuroinflammatory changes, including chronic microgliosis are key pathological components of AD. Microglia, the resident immune cells of the brain, is constantly survey the microenvironment under physiological conditions. In AD, deposition of β-amyliod (Aβ) peptide initiates a spectrum of cerebral neuroinflammation mediated by activating microglia. Activated microglia may play a potentially detrimental role by eliciting the expression of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α) influencing the surrounding brain tissue. This review will discuss the mechanisms and important role of pro-inflammatory cytokines in the pathogenesis of AD, and the ongoing drug targeting pro-inflammatory cytokine for therapeutic modulation.
Willette, A. A., Johnson, S. C., Birdsill, A. C., Sager, M. A., Christian, B., Baker, L. D., et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s Dem. 2015;11:504–510.
This is the first human study to demonstrate that insulin resistance may contribute to amyloid deposition in brain regions affected by AD.
Xu M-Q, Cao H-L, Wang W-Q, Wang S, Cao X-C, Yan F, et al. Fecal microbiota transplantation broadening its application beyond intestinal dosrders. World J Gastroenterol. 2015 Jan 7;21(1):102-111.
Intestinal dysbiosis is now known to be a complication in a myriad of diseases. Fecal microbiota transplantation (FMT), as a microbiota-target therapy, is arguably very effective for curing Clostridium difficile infection and has good outcomes in other intestinal diseases. New insights have raised an interest in FMT for the management of extra-intestinal disorders associated with gut microbiota. This review shows that it is an exciting time in the burgeoning science of FMT application in previously unexpected areas, including metabolic diseases, neuropsychiatric disorders, autoimmune diseases, allergic disorders, and tumors. A randomized controlled trial was conducted on FMT in metabolic syndrome by infusing microbiota from lean donors or from self-collected feces, with the resultant findings showing that the lean donor feces group displayed increased insulin sensitivity, along with increased levels of butyrate-producing intestinal microbiota. Case reports of FMT have also shown favorable outcomes in Parkinson’s disease, multiple sclerosis, myoclonus dystonia, chronic fatigue syndrome, and idiopathic thrombocytopenic purpura. FMT is a promising approach in the manipulation of the intestinal microbiota and has potential applications in a variety of extra-intestinal conditions associated with intestinal dysbiosis.
Yabu T., Shiba H., Shibasaki Y., Nakanishi T., Imamura S., Touhata K., et al. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015;22:258–273.
Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell Springer. 2015;6:254–64.
Akbari E, Asemi Z, Daneshvar Kakhaki R, et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: a randomized, double-blind and controlled trial. Front Aging Neurosci. 2016;8:256.
Alzheimer's disease (AD) is associated with severe cognitive impairments as well as some metabolic defects. Scant studies in animal models indicate a link between probiotics and cognitive function. This randomized, double-blind, and controlled clinical trial was conducted among 60 AD patients to assess the effects of probiotic supplementation on cognitive function and metabolic status. After 12 weeks intervention, compared with the control group, the probiotic treated patients showed a significant improvement in the MMSE score (P <0.001). Overall, the current study demonstrated that probiotic consumption for 12 weeks positively affects cognitive function and some metabolic statuses in the AD patients.
Anrather J, Ladecola C. Inflammation and stroke: an overview. Neurotherapeutics. 2016Oct;13(4):661-670.
The immune response to acute cerebral ischemia is a major factor in stroke pathobiology and outcome. While the immune response starts locally in occluded and hypoperfused vessels and the ischemic brain parenchyma, inflammatory mediators generated propagate through the organism as a whole. In this overview we will outline the inflammatory cascade from its starting point in the vasculature of the ischemic brain to the systemic immune response elicited by brain ischemia. Potential immunomodulatory therapeutic approaches, will also be discussed.
Bellan M, Pirisi M, Sainaghi PP. The Gas6/TAM System and Multiple Sclerosis. Int J Mol Sci. 2016 Nov;17(11):807.
Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016 Jun;12(6):719-32.
Several attempts have been made to treat Alzheimer's disease (AD) using anti-amyloid strategies with disappointing results. It is clear that the "amyloid cascade hypothesis" alone cannot fully explain the neuronal damage in AD, as evidenced both by autopsy and imaging studies. Neuroinflammation plays a significant role in neurodegenerative diseases, whereas the debate is ongoing about its precise role, whether it is protective or harmful. In this review, we focus on the potential mechanism of glial activation and how local and systemic factors influence disease progression. We focus on neuroinflammation in AD, especially in the earliest stages.
Elobeid A, Libard S, Leino M, Popova SSN, Alafuzoff I. Altered proteins in the aging brain. J Neuropathol Exp Neurol. 2016;75:316-25.
Ferland G, Feart C, Presse N, Lorrain S, Bazin F, Helmer C, et al. Vitamin K antagonists and cognitive function in older adults: The Three-City cohort study. J Gerontol A Biol Sci Med Sci. 2016 Oct;71(10:1356-1362.
A growing body of evidence supports a beneficial role for vitamin K in brain and cognition, notably in studies where animals are rendered vitamin K deficient by warfarin, a potent vitamin K antagonist (VKA). Given VKAs are commonly used oral anticoagulants in older persons, we investigated the relationship between VKA therapy and cognitive performances over 10 years in participants of the Three-City study. Treatment with VKAs was not associated with global cognitive functioning neither with rate of subsequent decline in scores on all three cognitive tests. No associations were found between platelet aggregation inhibitors and cognitive performances or rate of decline. These findings do not indicate a long-term detrimental effect of VKAs on cognition, but the risk–benefit balance of VKA treatment still deserves further research.
Fjell A.M., Sneve M.H., Storsve A.B., Grydeland H., Yendiki A., Walhovd K.B. Brain events underlying episodic memory changes in aging: A longitudinal investigation of structural and functional connectivity. Cereb. Cortex. 2016;26:1272–1286.
Fourgeaud, L., Través, P. G., Tufail, Y., Leal-Bailey, H., Lew, E. D., Burrola, P. G., et al. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532:240–244.
Garcia-Caceres C, Quarta ., Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016;166:–880.
Healy LM, Perron G, Won S-Y, Michell-Robinson MA, Rezk A, Ludwin S. K, et al. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J. Immunol. 2016;196:3375–3384.
Kapasi A, Schneider JA. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim Biophys Acta. 2016 May;1862(5):878-86.
There is growing evidence suggesting that vascular pathologies and dysfunction play a critical role in cognitive impairment, clinical Alzheimer's disease, and dementia. Vascular pathologies such as macroinfarcts, microinfarcts, microbleeds, small and large vessel cerebrovascular disease, and white matter disease are common especially in the brains of older persons where they contribute to cognitive impairment and lower the dementia threshold. Vascular dysfunction resulting in decreased cerebral blood flow, and abnormalities in the blood brain barrier may also contribute to the Alzheimer's disease (AD) pathophysiologic process and AD dementia. This review provides a clinical-pathological perspective on the role of vessel disease, vascular brain injury, alterations of the neurovascular unit, and mixed pathologies in the Alzheimer's disease pathophysiologic process and Alzheimer's dementia.
Kim SY, Lim EJ, Yoon YS, Ahn YH, Park EM, Kim HS, Kang JL. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci. Rep. 2016;6:1–16.
Kıray H, Lindsay SL, Hosseinzadeh S, Barnett SC. The multifaceted role of astrocytes in regulating myelination. Exp Neurol. 2016:541–9.
McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimer’s Dis 2016;54(3):853–57.
Two basic discoveries spurred research into inflammation as a driving force in the pathogenesis of Alzheimer's disease (AD). The first was the identification of activated microglia in association with the lesions. The second was the discovery that rheumatoid arthritics, who regularly consume anti-inflammatory agents, were relatively spared from the disease. More than 15 epidemiological studies have since showed a sparing effect of non-steroidal anti-inflammatory drugs (NSAIDs) in AD. A consistent finding has been that the longer the NSAIDs were used prior to clinical diagnosis, the greater the sparing effect. The reason has since emerged from studies of biomarkers such as amyloid-β (Aβ) levels in the cerebrospinal fluid and Aβ deposits in brain. They have established that the onset of AD commences at least a decade before cognitive decline permits clinical diagnosis. Neuroinflammation, discovered in AD more than 30 years ago, has now become a major field of brain research today. Inhibiting it may be the key to successful treatment of many chronic neurological disorders.
Minter MR, Zhang C, Leone V, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep. 2016;6:30028.
Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol 2010;6:193-201.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016 Aug 19;353(6301):777-83.
Neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and frontotemporal lobar dementia are among the most pressing problems of developed societies with aging populations. Neurons carry out essential functions such as signal transmission and network integration in the central nervous system and are the main targets of neurodegenerative disease. In this Review, I address how the neuron's environment also contributes to neurodegeneration. Maintaining an optimal milieu for neuronal function rests with supportive cells termed glia and the blood-brain barrier. Accumulating evidence suggests that neurodegeneration occurs in part because the environment is affected during disease in a cascade of processes collectively termed neuroinflammation. These observations indicate that therapies targeting glial cells might provide benefit for those afflicted by neurodegenerative disorders.
Redmann M., Darley-Usmar V., Zhang J., The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: Implications for neurodegenerative diseases. Aging Dis. 2016;7:150–162.
Soutif-Veillon A, Ferland G, Rolland Y, Presse N, Boucher K, Feart C, et al. Increased dietary vitamin K intake is associated with less severe subjective memory complaint among older adults. Maturitas. 2016;92:131-136.
Increased dietary intake of vitamin K, a fat-soluble nutrient involved in brain health and function, has been associated with better cognitive performance in older adults. Our objective was to determine whether the dietary vitamin K intake was associated with the presence and severity of subjective memory complaint among older adults. The results showed that increased dietary vitamin K intake was associated with fewer and less severe subjective memory complaint in older adults taking no vitamin K antagonists. These findings provide epidemiological data supporting future vitamin K replacement trials.
Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016 Apr;139(Pt 4):1265-81.
In addition to amyloid-β plaque and tau neurofibrillary tangle deposition, neuroinflammation is considered a key feature of Alzheimer's disease pathology. Inflammation in Alzheimer's disease is characterized by the presence of reactive astrocytes and activated microglia surrounding amyloid plaques, implicating their role in disease pathogenesis. In this study, we set out to determine if chronically activated microglia in the Alzheimer's disease brain are also dependent on CSF1R signalling, and if so, how these cells contribute to disease pathogenesis. Collectively, these results demonstrate that microglia contribute to neuronal loss, as well as memory impairments in mice, but do not mediate or protect from amyloid pathology.
Sterling NW, Lichtenstein M, Lee EY, Lewis MM, Evans A, Eslinger PJ, et al. Higher plasma LDL-cholesterol is associated with preserved executive and fine motor functions in Parkinson’s disease. Aging Dis. 2016;7:237–245.
Verkhratsky A, Matteoli M, Parpura V, Mothet J-P, Zorec R. Astrocytes as secretory cells of the central nervous system; idiosyncrasies of vesicular secretion. EMBO J. 2016 Feb 1;35(3):239-57.
Astrocytes are housekeepers of the central nervous system (CNS) and are important for CNS development, homeostasis and defenses. They communicate with neurons and other glial cells through the release of signaling molecules. Astrocytes secrete a wide array of classic neurotransmitters, neuromodulators and hormones, as well as metabolic, trophic and plastic factors, all of which contribute to the gliocrine system. In this review, we summarize the features and functions of secretory organelles in astrocytes. We focus on the biogenesis and trafficking of secretory organelles and on the regulation of the exocytotic secretory system in the context of healthy and diseased astrocytes.
Visioli F, Burgos-Ramos E. Selected micronutrients in cognitive decline prevention and therapy. Emol Neurobio. 2016 Aug;53(6):4083-4093.
The important role of diets and healthy lifestyle as preventative of neurodegenerative diseases is widely accepted nowadays, and it may provide preventive strategies in very early, non-symptomatic phases of dementia well, especially because there are still no effective treatments for it. In this article, we review the known effects of selected micronutrients on the aging brain and we propose strategies for dietary improvements
Yu Y-x, Li Y-P, Gao F, Hu Q-s, Zhang Y, Chen G, et al. Vitamin K2 suppresses rotenone-induced microglial activation in vitro. Acta Pharmacologica Sinica. 2016;37:1178-89.
Increasing evidence has shown that environmental factors such as rotenone and paraquat induce neuroinflammation, which contributes to the pathogenesis of Parkinson's disease (PD). In this study, we investigated the molecular mechanisms underlying the repression by menaquinone-4 (MK-4), a subtype of vitamin K2, of rotenone-induced microglial activation in vitro. We found that vitamin K2 can directly suppress rotenone-induced activation of microglial BV2 cells in vitro by repressing ROS production.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53.]
Zhu YZ, Wang W, Xian N, Wu B. Inhibition of TYRO3/Akt signaling participates in hypoxic injury in hippocampal neurons. Neural Regen Res. (2016) 11:752–7.
Aarsland D, Creese B, Politis M, Chaudhuri KR, Ffytche DH, Weintraub D, Ballard C. Cognitive decline in parkinson disease. Nat. Rev. Neurol. 2017;13:217–231.
Abboud-Jarrous G, Priya S, Maimon A, Fischman S, Cohen-Elisha M, Czerninski R, et al. Protein S drives oral squamous cell carcinoma tumorigenicity through regulatin of AXL. Oncotarget. 2017 Feb 21;8(8):13986-14002.
The TAM family, comprising of TYRO3, AXL, and MERTK, is implicated in many human cancers. Their activation leads to cancer cell proliferation, enhanced migration, invasion, and drug resistance; however how TAMs are activated in cancers is less understood. We previously showed that Protein S (PROS1) is a ligand of the TAM receptors. Here we identify PROS1 as a mediator of Oral Squamous Cell Carcinoma (OSCC) in proliferation, cell survival and migration. We demonstrate that excess PROS1 induces OSCC proliferation and migration. Taken together, we identify PROS1 as a driver of OSCC tumor growth and a modulator of AXL expression. Our results point to PROS1 as a potential novel anti-cancer therapeutic target.
Bondi MW, Edmonds EC, Salmon DP. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017;23:818–831.
Although dementia has been described in ancient texts over many centuries (e.g., “Be kind to your father, even if his mind fail him.” – Old Testament: Sirach 3:12), our knowledge of its underlying causes is little more than a century old. Alzheimer published his now famous case study only 110 years ago, and our modern understanding of the disease that bears his name, and its neuropsychological consequences, really only began to accelerate in the 1980s. We review this lineage of work beginning with Alzheimer’s own writings and drawings, then jump to the modern era beginning in the 1970s and early 1980s and provide a sampling of neuropsychological and other contextual work from each ensuing decade. Finally, we suggest future directions and predictions for dementia-related research and potential therapeutic interventions.
Bruce KD, Zsombok A, Eckel RH. Lipid processing in the brain: a key regulator of systemic metabolism. Front Endocrinol. 2017 Apr; https://doi.org/10.3389/fendo.2017.00060
Burstyn-Cohen T . TAM receptor signaling in development. Vol. 61, International Journal of Developmental Biology. 2017. pp. 215-24.
Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017 Jan;49:60-68.
The pathway leading from amyloid-β deposition to cognitive impairment is believed to be a cornerstone of the pathogenesis of Alzheimer's disease (AD). However, what drives amyloid buildup in sporadic nongenetic cases of AD is still unknown. AD brains feature an inflammatory reaction around amyloid plaques, and a specific subset of the gut microbiota (GMB) may promote brain inflammation. We investigated the possible role of the GMB in AD pathogenesis Our data indicate that an increase in the abundance of a pro-inflammatory gut microbiota taxon, Escherichia/Shigella, and a reduction in the abundance of an anti-inflammatory taxon, E. rectale, are possibly associated with a peripheral inflammatory state in patients with cognitive impairment and brain amyloidosis. A possible causal relation between GMB-related inflammation and amyloidosis deserves further investigation.
Chen D, Yang X, Yang J, Lai G, Yong T, Tang X, Shuai O, et al. Prebiotic effect of Fructooligosaccharides from Morinda Officinalis on Alzheimer’s disease in roden models by targeting the microbiota-gut-brain axis. Front Aging Neurosci. 2017 Dec 8;9:403.
Gut microbiota influences the central nervous system disorders such as Alzheimer's disease (AD). The prebiotics and probiotics can improve the host cognition. A previous study demonstrated that fructooligosaccharides from Morinda officinalis (OMO) exert effective memory improvements in AD-like animals, thereby considered as potential prebiotics; however, the underlying mechanism still remains enigma. Thus, the present study investigated whether OMO is effective in alleviating AD by targeting the microbiota-gut-brain axis. Taken together, our findings suggest that the therapeutic effect of the traditional medicine, M. officinalis, on various neurological diseases such as AD, is at least partially contributed by its naturally occurring chemical constituent, OMO, via modulating the interaction between gut ecology and brain physiology.
Downey CL, Young A, Burton EF, Graham SM, Macfarlane RJ, Tsapakis EM, et al. Dementia and osteoporosis in a geriatric population: is there a common link? World J Orthop. 2017;8:412-423.
Garcia-Pena C, Alvarez-Cisneros T, Quiroz-Baez R, Friedland RP. Microbiota and aging: a review and commentary. Arch Med Res. 2017 Nov;48(8):681-89.
Although there is a consensus that the dominant species that make up the adult microbiota remains unchanged in elderly people, it has been reported that there are significant alterations in the proportion and composition of the different taxa, leading to reduced microbiota diversity, as well as an increase of enteropathogens that may lead to chronic inflammation. The ageing of mucosal immune and motor systems also contributes to these changes. Also, environment, diet, place of residence and biogeography are regulatory factors of the microbiota. Communication in the gut-brain-axis is regulated by many intermediaries including diverse metabolites of the microbiota. Microbial changes have been observed in several geriatric diseases, like Parkinson's and Alzheimer's. In addition, evidence has shown that individuals with high frailty scores had a significant reduction on lactobacilli species when compared to non-frail individuals. Oral microbiota may be also especially important because of the opportunities for access to the brain through the olfactory nerve at the roof of the nose or through the abundant innervations of the oral cavity by the trigeminal and other cranial nerves. The study of the microbiota represents an important advance in the understanding of the aging process.
Harach, T., Marungruang, N., Duthilleul, N., Cheatham, V., Mc Coy, K. D., Frisoni, G., et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017;7:41802.
Jäkel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Frontiers in Cellular NeuroSci. 2017; https://doi.org/10.3389/fncel.2017.00024
Glial cells, consisting of microglia, astrocytes, and oligodendrocyte lineage cells as their major components, constitute a large fraction of the mammalian brain. Originally considered as purely non-functional glue for neurons, decades of research have highlighted the importance as well as further functions of glial cells. Although many aspects of these cells are well characterized nowadays, the functions of the different glial populations in the brain under both physiological and pathological conditions remain, at least to a certain extent, unresolved. In this review, we will give a detailed summary on different glial ablation studies, focusing on the adult mouse central nervous system and the functional readouts. We will also provide an outlook on how these approaches could be further exploited in the future.
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017; 40:151–166
Khrimian L, Obri A, Ramos-Brossier M, Rousseaud A, Moriceau S, Nicot AS, et al. Gpr158 mediates osteocalcin's regulation of cognition. J Exp Med. 2017 Oct 2; 214(10):2859-2873.
Kimura K, Hirota Y, Kuwahara S, Takeuchi A, Tode C, Wada A, et al. Synthesis of Novel Synthetic Vitamin K Analogues Prepared by Introduction of a Heteroatom and a Phenyl Group That Induce Highly Selective Neuronal Differentiation of Neuronal Progenitor Cells. J Med Chem. 2017 Mar 23; 60(6):2591-2596.
Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, et al. Head injury as a risk factor for dementia and Alzheimer’s disease: A systematic review and meta-analysis of 32 observational studies. PLoS One. 2017;12(1).
The findings from this meta-analysis indicate that head injury is associated with increased risks of dementia and AD.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017 Jan 26;541(7638):481-87.
Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention, and care. Lancet. 2017;390:2673–2734.
McManus R, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017 Mar 4;9:14.
The past 15 years have established a key role for infectious pathogens in the progression of age-related neurodegeneration. It is now accepted that cells of the central nervous system are sensitive to both the inflammatory events occurring in the periphery and to the infiltration of peripheral immune cells. This is particularly relevant for the progression of Alzheimer's disease, in which it has been demonstrated that patients are more vulnerable to infection-related cognitive changes. This review will discuss the impact of each of these infections, and examine the changes known to occur with age in the immune system.
Mullins, R. J., Diehl, T. C., Chia, C. W., and Kapogiannis, D. Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer's disease. Front. Aging Neurosci. 2017;9:118.
This article attempted to disentangle the complex mechanisms underlying brain insulin resistance (IR) highlight proven or plausible links to Aβ and tau pathologies in AD. The convergence of such diverse sources of evidence makes it all but certain that brain IR plays a major role in AD pathogenesis linking the two main types of pathology/. Ultimately, the merit of this hypothesis rests on demonstrating effectiveness in ongoing and future clinical trials.
Olsen AE, Færgeman NJ. Spingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017;7:170069.
Panagopoulos GN, Megaloikonomos PD, Mavrogenis AF. The present and future for peripheral nerve regeneration. Orthopedics. SLACK Incorporated. 2017;40:e141–56.
Ponziani FR, Pompili M, Di Stasio E, Zocco MA, Gasbarrini A, Flore R. Subclinical atherosclerosis is linked to small intestinal bacterial overgrowth via vitamin K2-dependent mechanisms. World J Gastroenterol. 2017;23:1241–1249.
SIBO is associated with reduced matrix Gla-protein activation as well as arterial stiffening. Both these observations are regarded as important indicators of subclinical atherosclerosis. Hence, screening for SIBO, intestinal decontamination and supplementation with vitamin K2 has the potential to be incorporated into clinical practice as additional preventive measures.
Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic Biol Med. 2017 Jan;102:203-16.
An important concept in neurobiology is "neurons that fire together, wire together" which means that the formation and maintenance of synapses is promoted by activation of those synapses. Very similar to the effects of the stress of exercise on muscle cells, emerging findings suggest that neurons respond to activity by activating signaling pathways that stimulate mitochondrial biogenesis and cellular stress resistance. These pathways are also activated by aerobic exercise and food deprivation. The metabolic 'switch' in fuel source from liver glycogen store-derived glucose to adipose cell-derived fatty acids and their ketone metabolites during fasting and sustained exercise, appears to be a pivotal trigger that enhance learning and memory and underlying synaptic plasticity and neurogenesis. Emerging findings suggest that fasting, exercise and an intellectually challenging lifestyle can protect neurons against the dysfunction and degeneration that they would otherwise suffer in acute brain injuries (stroke and head trauma) and neurodegenerative disorders including Alzheimer's, Parkinson's and Huntington's disease. A better understanding of such fundamental neuronal response mechanisms is expected to result in the development and implementation of novel interventions to promote optimal brain function and healthy brain aging.
Ray AK, DuBois JC, Gruber RC, Guzik HM, Gulinello ME, Perumal G, et al. Loss of Gas6 and Axl signaling results in extensive axonal damage, motor deficits, prolonged neuroinflammation and less remyelination following cuprizone exposure. Glia. 2017 Dec;65(12):2015-2069.
The TAM (Tyro3, Axl, and MerTK) family of receptor tyrosine kinases (RTKs) and their ligands, Gas6 and ProS1, are important for innate immune responses and central nervous system (CNS) homeostasis. While only Gas6 directly activates Axl, ProS1 activation of Tyro3/MerTK can indirectly activate Axl through receptor heterodimerization. Therefore, we generated Gas6−/−Axl−/− double knockout (DKO) mice to specifically examine the contribution of this signaling axis while retaining ProS1 signaling through Tyro3 and MerTK. We found that the Gas6-Axl signaling plays an important role in maintaining axonal integrity and regulating and reducing CNS inflammation that cannot be compensated for by ProS1/Tyro3/MerTK signaling.
Ricciarelli R, Fedele E. The Amyloid C\cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr Neuropharmacol. 2017 Aug;15(6):926-935.
Since its discovery in 1984, the beta amyloid peptide has treaded the boards of neurosciences as the star molecule in Alzheimer’s disease pathogenesis. In the last decade, however, this vision has been challenged by evidence-based medicine showing the almost complete failure of clinical trials that experimented anti-amyloid therapies with great hopes. Moreover, data have accumulated which clearly indicate that this small peptide plays a key role in the physiological processes of memory formation. In the present review, we will discuss the different aspects of the amyloid cascade hypothesis, highlighting its pros and cons, and we will analyze the results of the therapeutic approaches attempted to date that should change the direction of Alzheimer’s disease research in the future.
Ridone P, Grage SL, Patkunarajah A, Battle AR, Ulrich AS, Martinac B. “Force-from-lipids” gating of mechanosensitive channels modulated by PUFAs. J Mech Behav Biomed Mater Netherlands. 2017;79:158–67.
Sanabria-Castro A, Alvarado-Echeverra I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci. 2017 May;24(1):46-54.
Dementia is a chronic or progressive syndrome, characterized by impaired cognitive capacity beyond what could be considered a consequence of normal aging. It affects the memory, thinking process, orientation, comprehension, calculation, learning ability, language, and judgment; although awareness is usually unaffected. Alzheimer's disease (AD) is the most common form of dementia; symptoms include memory loss, difficulty solving problems, disorientation in time and space, among others. This literature review summarizes the main underlying neurobiological mechanisms in AD, including the theory with emphasis on amyloid peptide, cholinergic hypothesis, glutamatergic neurotransmission, the role of tau protein, and the involvement of oxidative stress and calcium.
Schwalfenberg G.K. Vitamins K1 and K2: The emerging group of vitamins required for human health. J. Nutr. Metab. 2017;2017:6254836.
Vitamin K2 may be a useful adjunct for the treatment of osteoporosis, along with vitamin D and calcium, rivaling bisphosphonate therapy without toxicity. It may also significantly reduce morbidity and mortality in cardiovascular health by reducing vascular calcification. Vitamin K2 appears promising in the areas of diabetes, cancer, and osteoarthritis. Vitamin K use in warfarin therapy is safe and may improve INR control, although a dosage adjustment is required. Vitamin K supplementation may be useful for a number of chronic conditions that are afflicting North Americans as the population ages. Supplementation may be required for bone and cardiovascular health.
Wang J, G BJ, Masters CL, Wang Y-J. A systemic view of Alzheimer disease-insights from amyloid- β beyond metabolism beyond the brain. Nat Rev Neurol. 2017 Sep 29;13(10):612-23.
Alzheimer disease (AD) is the most common type of dementia, and is currently incurable; existing treatments for AD produce only a modest amelioration of symptoms. Research into this disease has conventionally focused on the CNS. However, several peripheral and systemic abnormalities are now understood to be linked to AD, and our understanding of how these alterations contribute to AD is becoming more clearly defined. This Review focuses emerging findings of associations between systemic abnormalities and Aβ metabolism, and describe how these associations might interact with or reflect on the central pathways of Aβ production and clearance. On the basis of these findings, we propose that these abnormal systemic changes might not only develop secondary to brain dysfunction but might also affect AD progression, suggesting that the interactions between the brain and the periphery have a crucial role in the development and progression of AD.
Westfall S, Lomis N, Kahouli I, Dia SY, Singh SP, Prakash S. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell Mol Life Sci. 2017;74:3769-3787.
The gut microbiota is essential to health and has recently become a target for live bacterial cell biotherapies for various chronic diseases including metabolic syndrome, diabetes, obesity and neurodegenerative disease. Probiotic biotherapies are known to create a healthy gut environment by balancing bacterial populations and promoting their favorable metabolic action. The microbiota and its respective metabolites communicate to the host through a series of biochemical and functional links thereby affecting host homeostasis and health. In particular, the gastrointestinal tract communicates with the central nervous system through the gut–brain axis to support neuronal development and maintenance while gut dysbiosis manifests in neurological disease. There are three basic mechanisms that mediate the communication between the gut and the brain: direct neuronal communication, endocrine signaling mediators and the immune system. Together, these systems create a highly integrated molecular communication network that link systemic imbalances with the development of neurodegeneration including insulin regulation, fat metabolism, oxidative markers and immune signaling. Age is a common factor in the development of neurodegenerative disease and probiotics prevent many harmful effects of aging such as decreased neurotransmitter levels, chronic inflammation, oxidative stress and apoptosis—all factors that are proven aggravators of neurodegenerative disease. Indeed patients with Parkinson’s and Alzheimer’s diseases have a high rate of gastrointestinal comorbidities and it has be proposed by some the management of the gut microbiota may prevent or alleviate the symptoms of these chronic diseases.
Wolf SA, Boddeke HWGM, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619-43.
As the immune-competent cells of the brain, microglia play an increasingly important role in maintaining normal brain function. They invade the brain early in development, transform into a highly ramified phenotype, and constantly screen their environment. Microglia are activated by any type of pathologic event or change in brain homeostasis. This activation process is highly diverse and depends on the context and type of the stressor or pathology. They are the professional phagocytes of the brain and help orchestrate the immunological response by interacting with infiltrating immune cells. We describe here the diversity of microglia phenotypes and their responses in health, aging, and disease.
Zelentsova K, Talmi Z, Abboud-Jarrous G, Sapir T, Capucha T, Nassar M, et al. Protein S Regulates Neural Stem Cell Quiescence and Neurogenesis. Stem Cells. 2017;35(3):679–93.
Neurons are continuously produced in brains of adult mammalian organisms throughout life-a process tightly regulated to ensure a balanced homeostasis. In the adult brain, quiescent Neural Stem Cells (NSCs) residing in distinct niches engage in proliferation, to self-renew and to give rise to differentiated neurons and astrocytes. The mechanisms governing the intricate regulation of NSC quiescence and neuronal differentiation are not completely understood. Here, we report the expression of Protein S (PROS1) in adult NSCs, and show that genetic ablation of Pros1 in neural progenitors increased hippocampal NSC proliferation by 47%. We show that PROS1 regulates the balance of NSC quiescence and proliferation, also affecting daughter cell fate. Our study identifies a duple role for PROS1 in stem-cell quiescence and as a pro-neurogenic factor, and highlights a unique segregation of increased stem cell proliferation from enhanced neuronal differentiation, providing important insight into the regulation and control of NSC quiescence and differentiation.
Zuo H, Wang R, Jiang D, Fang D. Determining the composition of active C cholesterol in the plasma membrane of single cells by using electrochemiluminescence. Chem Electro Chem. 2017;4:1677–80.
Aguo X-Z, Shan C, Hou Y-F, Zhu G, Tao B, Sun L-H, et al. Osteocalcin ameliorates motor dysfunction in a 5-Hydroxydopamine-induced Parkinson’s disease rat model through AKT/GSK3β signaling. Front Mol Neurosci. 2018;11:343.
In summary, OCN plays a protective role against parkinsonian neurodegeneration in the PD rat model, suggesting a potential therapeutic use of OCN in PD.
Athari Nik Azm S, Djazayeri A, Safa M, et al. Lactobacillus and Bifidobacterium ameliorate memory and learning deficits and oxidative stress in Aβ (1–42) injected rats. Appl Physiol Nutr Metab. 2018;43:718–726.
The gastrointestinal microbiota affects brain function, including memory and learning. In this study we investigated the effects of probiotics on memory and oxidative stress biomarkers in an experimental model of Alzheimer's disease. The Alzheimer-probiotics group had significantly improved spatial memory, including shorter escape latency and travelled distance and greater time spent in the target quadrant. There was also improvement in oxidative stress biomarkers such as increased malondialdehyde levels and superoxide dismutase activity following the β-amyloid injection. Overall, it seems that probiotics play a role in improving memory deficit and inhibiting the pathological mechanisms of Alzheimer's disease by modifying microbiota.
Brangier A, Celle S, Roche F, Beauchet O, Ferland G, Annweiler C. Use of vitamin K antagonists and brain morphological changes in older adults: an exposed/unexposed voxel-based morphometric study. Dement Geriatr Cogn Disord. 2018;45(1–2):18–26.
Vitamin K antagonists (VKAs) are commonly used for their role in haemostasis by interfering with the vitamin K cycle. Since vitamin K also participates in brain physiology, this voxel-based morphometric study aimed to determine whether the duration of exposure to VKAs correlated with focal brain volume reduction in older adults. They found focal atrophies in older adults exposed to VKA. These findings provide new insights elucidating the effects of VKAs on brain health and function in older adults.
Brangier A, Ferland G, Rolland Y, Gautier J, Feart C, Annweiler C. Vitamin K antagonists and cognitive decline in older adults: a 24-month follow-up. Nutrients. 2018;10(6):E666.
Vitamin K participates in brain physiology. This study aimed to determine whether using vitamin K antagonists (VKAs), which interfere with the vitamin K cycle, were (i) cross-sectionally associated with altered cognitive performance, and (ii) independent predictors of cognitive changes in older adults over 24 months. In conclusion, we found more severe executive dysfunction at baseline and incident executive decline over 24 months among geriatric patients using VKAs, when compared with their counterparts.
Catapano AL. Dyslipidaemias in 2017: Atherogenic lipoproteins as treatment targets. Nat Rev Cardiol. 2018;15:75–6.
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204–18.
Chung J, Phukan G, Vergote D, Mohamed A, Maulik M, Stahn M, et al. Endosomal-lysosomal cholesterol sequestration by U18666A differentially regulates APP metabolism in normal and APP overexpressing cells. Mol Cell Biol. 2018;MCB.00529–17.
Cui C, Sekikawa K, Kuller LH, Lopez OL, Newman AB, Kuipers AL, et al. Aortic stiffness is associated with increased risk of incident dementia in older adults. J Alzheimers Dis. 2018;66(1):297-306.
Cardiovascular disease risk factors, including age, hypertension, and diabetes, contribute to aortic stiffness and subclinical cardiovascular and brain disease, increasing dementia risk. Aortic stiffness, measured by carotid-femoral pulse wave velocity (cfPWV), reduces the buffering of pulsatile blood flow, exposing cerebral small arteries to microvascular damage. High cfPWV is related to white matter hyperintensities and brain amyloid deposition, and to cognitive decline. This study looked at the hypothesis that cfPWV predicts incident dementia in older adults, independent of potential confounders. They found that cfPWV was significantly associated with increased risk of dementia, but systolic blood pressure, mean arterial pressure and pulse pressure were not. Finally, higher cfPWV was related to lower physical activity intensity and higher systolic blood pressure, heart rate, and waist circumference measured 5 years prior. An important unanswered question is whether interventions to slow arterial stiffening can reduce the risk of dementia.
Dahlback B. Vitamin K-dependent protein S: beyond the protein C pathway. Semin Thromb Hemost. 2018 Mar;44(2):176-184.
Protein S is a vitamin K-dependent plasma glycoprotein circulating in plasma at a concentration of around 350 nM. Protein S in plasma has multiple anticoagulant properties and heterozygous protein S deficiency is associated with increased risk of venous thrombosis. Protein S has additional functions beyond being an anticoagulant. Protein S interacts with tyrosine kinase receptors of the TAM family, which comprises Tyro3, Axl, and Mer. The TAM receptor interaction is important for the ability of protein S to stimulate phagocytosis of apoptotic cells. This review will discuss the multiple functions of protein S, describing its role as cofactor to activated protein C with a subsequent focus on the other functions of protein S.
Ekblad, LL, Johansson J, Helin S, Viitanen M, Laine H, Puukka P, et al. Midlife insulin resistance. APOE genotype, and late-life brain amyloid accumulation. Neurology. 2018;90:e1150–e1157.
These results indicate that midlife insulin resistance is an independent risk factor for brain amyloid accumulation in elderly individuals without dementia.
Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358.
Neurodegenerative diseases have two general characteristics that are so fundamental we usually take them for granted. The first is that the pathology associated with the disease only affects particular neurons (‘selective neuronal vulnerability’); the second is that the pathology worsens with time and impacts more regions in a stereotypical and predictable fashion. Here we detail our emerging understanding of the underlying biology of selective neuronal vulnerability and outline some of the areas in which our understanding is incomplete.
Guo XZ, Shan C, Hou YF, Zhu G, Tao B, Sun LH, et al. Osteocalcin ameliorates motor dysfunction in a 6-Hydroxydopamine-induced Parkinson's disease rat model through AKT/GSK3beta signaling. Front Mol Neurosci. 2018;11(343):10.3389/fnmol.
Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nature Reviews Mole Cell Biol. 2018;19:175-191.
Studies of bioactive lipids in general and sphingolipids in particular have intensified over the past several years, revealing an unprecedented and unanticipated complexity of the lipidome and its many functions, which rivals, if not exceeds, that of the genome or proteome. These results highlight critical roles for bioactive sphingolipids in most, if not all, major cell biological responses, including all major cell signaling pathways, and they link sphingolipid metabolism to key human diseases.
Hussain G, Rasul A, Anwar H, Aziz N, Razzaq A, Wei W, et al. Role of plant derived alkaloids and their mechanism in neurodegenerative disorders. Int J Biol Sci. 2018;14:341–57.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia 2018;4:575-590.
Alzheimer’s disease is a progressive neurodegenerative disorder that is characterized by cognitive decline and the presence of two core pathologies, amyloid β plaques and neurofibrillary tangles. Over the last decade, the presence of a sustained immune response in the brain has emerged as a third core pathology in AD. The sustained activation of the brain’s resident macrophages (microglia) and other immune cells has been demonstrated to exacerbate both amyloid and tau pathology and may serve as a link in the pathogenesis of the disorder. In the following review, we provide an overview of inflammation in AD and a detailed coverage of a number of microglia-related signaling mechanisms that have been implicated in AD. We also review the potential connection of risk factors for AD and how they may be related to inflammatory mechanisms.
Kodis EJ, Choi S, Swanson E, Ferreira G, Bloom GS. N-methyl-D-aspartate receptor–mediated calcium influx connects amyloid-β oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimers Dement. 2018;14:1302–12.
Lasemi R, Kundi M, Moghadam NB, Moshammer H, Hainfellner JA. Vitamin K2 in multiple sclerosis patients. Wien Klin Wochenschr. 2018 May; 130(9-10):307-313.
Due to its role in protection of mitochondrial damage, VK2 could be relevant in preventing disease progress in multiple sclerosis (MS). The MS patients had vastly lower VK2 blood levels than controls (235 ± 100 ng/ml vs. 812 ± 154 ng/ml, respectively). Female patients had significantly lower VK2 levels than males and a decrease with age by approximately 10% per decade was found. The substantially lower levels of VK2 in MS patients could be due to depletion, lower production in the gut, diminished absorption or, less likely, reduced intake of precursor vitamin K1. The role of VK2 in MS development and progress deserves further study.
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757-772.
Lu MH, Zhao XY. Yao PP, Xu DE, Ma QH. The mitochondrion: a potential therapeutic target for Alzheimer’s disease. Neurosci. Bull. 2018;34:1127–1130.
The mitochondrion is the “energy plant” that provides most of the energy for cells. Mitochondria also participate in various processes such as calcium homeostasis, cell death, and cell growth during development and aging. Mitochondrial abnormalities are a common phenomenon in aging and age-related neurodegenerative diseases such as Alzheimer’s disease (AD). Mitochondrial dysfunction is even taken to be a marker for aging, an indispensable risk factor for AD. Anti-oxidants and factors that maintain mitochondrial homeostasis have beneficial effects in AD therapy. Thus, the mitochondrion is emerging as a novel target for AD therapy.
Lumbroso D, Soboh S, Maimon A, Schif-Zuck S, Ariel A, Burstyn-Cohen T. Macrophage derived protein S facilitates apoptotic polymorphonuclear cell clearance by resolution phase macrophages and supports their reprogramming. Front Immunol. 2018;9:358.
Markopoulou K, Compta Y. Cerebrospinal fluid levels of alpha-synuclein in Parkinson's disease: another long and winding road. Park Relat Disord. 2018;49:1–3.
Morris MC, Wang Y, Barnes LL, Bennett DA, Dawson-Hughes B, Booth S. Nutrients and bioactives in green leafy vegetables and cognitive decline. Neurology. 2018;90(3): e214–e222.
To increase understanding of the biological mechanisms underlying the association, we investigated the individual relations to cognitive decline of the primary nutrients and bioactives in green leafy vegetables, including vitamin K (phylloquinone), lutein, β-carotene, nitrate, folate, kaempferol, and α-tocopherol. They found that higher intakes of each of the nutrients and bioactives except β-carotene were individually associated with slower cognitive decline. They concluded that consumption of approximately 1 serving per day of green leafy vegetables and foods rich in phylloquinone, lutein, nitrate, folate, α-tocopherol, and kaempferol may help to slow cognitive decline with aging. The identification of effective prevention strategies for dementia is critical to staving off a public health crisis and for meeting demand for this kind of information, particularly around diet. Among all of the different types of vegetables, green leafy vegetables have been identified as having the strongest protective relations against cognitive decline.
Moser SC, van der Eerden BCJ. Osteocalcin-A versatile bone-derived hormone. Front Endocrinol. 2018;9:794.
Bone has long been regarded as a static organ, simply providing protection and support. However, this mindset has changed radically in recent years and bone is becoming increasingly recognized for its endocrine function of secreting several hormones, thereby controlling various physiological pathways. One of the factors released by the skeleton is osteocalcin. Importantly, osteocalcin is secreted solely by osteoblasts but only has minor effects on bone mineralization and density. Instead, it has been reported to control several physiological processes in an endocrine manner, such as glucose homeostasis and exercise capacity, brain development, cognition, and male fertility. The aim of this review is to provide an overview of the currently known roles of osteocalcin and their underlying mechanisms.
Oveisgharan S, Buchman AS, Yu L, Farfel J, Hachinski V, Gaiteri C, et al. APOE ϵ2ϵ4 genotype, incident AD and MCI, cognitive decline, and AD pathology in older adults. Neurology. 2018;90:e2119–26.
To examine the association of the APOE ε2ε4 genotype with incident Alzheimer disease (AD), mild cognitive impairment (MCI), cognitive decline, and AD pathology in older adults. They concluded that APOE ε2ε4 genotype in older adults is associated with risk of MCI, cognitive decline, and a greater burden of AD pathology, especially β-amyloid.
Patterson, C. (2018). World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. London, UK: Alzheimer’s Disease International. Available online at: https://www.alz.co.uk/research/world-report-2018.
This report brings together 21 of the global leading lights in all areas of dementia research. Written by renowned journalist and broadcaster Christina Patterson (Time Magazine, The Guardian, The Sunday Times), this report tackles some of the complex questions surrounding dementia research. It looks at the hopes and frustrations and asks why there have been no major medical treatment breakthroughs for over 20 years. The report looks at a broad cross section of research areas including basic science; diagnosis; drug discovery; risk reduction and epidemiology. With the continued absence of a disease modifying treatment, the report also features progress, innovation and developments in care research.
Pizzorno L. Vitamin K2: optimal levels essential for the prevention of age-associated chronic disease. Longevity Med Rev. 2018;
Popescu DA, Huang H, Singhal NK, Shriver L, McDonough J, Clements RJ, et al. Vitamin K enhances the production of brain sulfatides during remyelination. PLOS ONE. 2018 Aug;13(8): e0203057
Multiple sclerosis (MS) is a devastating neurological disease, which is characterized by multifocal demyelinating lesions in the central nervous system. In this study we investigated the role of vitamin K in remyelination, by using an animal model for MS, the cuprizone model. Vitamin K enhanced the production of total brain sulfatides, after both 1 week and 3 weeks of remyelination, when compared with the control group. To determine whether or not there is a synergistic effect between vitamins K and D for the production of brain sulfatides, we employed a similar experiment as above. Vitamin K also increased the production of individual brain sulfatides, after 3 weeks of remyelination, when compared to the control group.
Popugaeva E, Pchitskaya E, Bezprozvanny I. Dysregulation of intracellular calcium signaling in Alzheimer’s disease. Antioxid Redox Signal United States. 2018;29:1176–88.
Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer’s disease. Rev Neurosci. 2018 Aug 28;29(6):613-27.
A major feature of Alzheimer's disease (AD) pathology is the plaque composed of aggregated amyloid-β (Aβ) peptide. Although these plaques may have harmful properties, there is much evidence to implicate soluble Aβ as the primary noxious form. Aβ is toxic to neurons in a myriad of ways. Current treatments for AD are limited and palliative. Much research and effort is being devoted to reducing Aβ production as an approach to slowing or preventing the development of AD. Aβ formation results from the amyloidogenic cleavage of human amyloid precursor protein (APP). Reconfiguring this process to disfavor amyloid generation might be possible through the reduction of APP or inhibition of enzymes that convert the precursor protein to amyloid.
Ross RD, Shah RC, Leurgans S, Bottiglieri T, Wilson RS, Sumner DR. Circulating Dkk1 and TRAIL are associated with cognitive decline in community-dwelling, older adults With cognitive concerns. J Gerontol A Biol Sci Med Sci. 2018 Nov 10;73(12):1688-1694.
Sanborn V, Azcarate-Peril MA, Updegraff J, Manderino LM, Gunstad J. A randomized clinical trial examining the impact of LGG probiotic supplementation on psychological status in middle-aged and older adults. Contemp Clin Trials Commun. 2018;12:192–197.
Shafit-Zagardo B, Gruber RC, and DuBois JC. The role of TAM family receptors and ligands in the nervous system: from development to pathobiology. Pharmacol. Ther. 2018;188:97–117.
Tyro3, Axl, and Mertk, referred to as the TAM family of receptor tyrosine kinases, are instrumental in maintaining cell survival and homeostasis in mammals. TAM receptors interact with multiple signaling molecules to regulate cell migration, survival, phagocytosis and clearance of metabolic products and cell debris called efferocytosis. The TAMs also function as rheostats to reduce the expression of proinflammatory molecules and prevent autoimmunity. All three TAM receptors are activated in a concentration-dependent manner by the vitamin K-dependent growth arrest-specific protein 6 (Gas6). Gas6 and the TAMs are abundantly expressed in the nervous system. Gas6, secreted by neurons and endothelial cells, is the sole ligand for Axl. ProteinS1 (ProS1), another vitamin K-dependent protein functions mainly as an anti-coagulant, and independent of this function can activate Tyro3 and Mertk, but not Axl. This review will focus on the role of the TAM receptors and their ligands in the nervous system.
Shang J, Yamashita T, Fukui Y, Song D, Li X, Zhai Y, et al. Different associations of plasma biomarkers in alzheimer’s disease, mild cognitive impairment, vascular dementia, and ischemic stroke. J Clin Neurol. 2018;14:29–34.
Strandwitz P. Neurotransmitter modulation by the gut microbiota. Pt Brain Res. 2018;1693:128–133.
The gut microbiota - the trillions of bacteria that reside within the gastrointestinal tract - has been found to not only be an essential component immune and metabolic health, but also seems to influence development and diseases of the enteric and central nervous system, including motility disorders, behavioral disorders, neurodegenerative disease, cerebrovascular accidents, and neuroimmune-mediated disorders. By leveraging animal models, several different pathways of communication have been identified along the "gut-brain-axis" including those driven by the immune system, the vagus nerve, or by modulation of neuroactive compounds by the microbiota. Of the latter, bacteria have been shown to produce and/or consume a wide range of mammalian neurotransmitters, including dopamine, norepinephrine, serotonin, or gamma-aminobutyric acid (GABA). Substantially more work is required to determine whether microbiota-mediated manipulation of human neurotransmission has any physiological implications, and if so, how it may be leveraged therapeutically. In this review this exciting route of communication along the gut-brain-axis, and accompanying data, are discussed.
Tamadon-Nejad S, Ouliass B, Rochford J, Ferland G. Vitamin K deficiency induced by warfarin is associated with cognitive and behavioral perturbations, and alterations in brain sphingolipids in rats. Front Aging Neurosi. 2018 Jul;10:213.
Initially discovered for its role in blood coagulation, there is now convincing evidence that vitamin K (VK) has important actions in the nervous system. In brain, VK is present in the form of menaquinone-4 (MK-4), a byproduct of the main dietary source, phylloquinone. It contributes to the biological activation of various proteins (i.e., Gas6), and participates in the synthesis of sphingolipids, a class of lipids widely present in brain cell membranes with important cell signaling functions. In a previous study, we reported that lifetime consumption of a low VK diet resulted in mild cognitive impairment in aged rats, a finding associated with an alteration of the sphingolipid profile. To confirm the role of VK as it relates to sphingolipids, cognition, and behavior outside the context of aging, we conducted a study of acute VK deficiency using a pharmacological model of VK deficiency in brain. The WVK treatment resulted in a dramatic decrease in MK-4 level in all brain regions despite the presence of high local concentrations of phylloquinone, Additionally, WVK treatment affected sphingolipid concentrations in key brain regions, notably those of the ganglioside family. Finally, brain MK-4 was correlated with performances in the open field test. This study confirms the modulatory role of VK in cognition and behavior and the implication of sphingolipids.
Trayssac, M., Hannun, Y. A., and Obeid, L. M. Role of Sphingolipids in senescence: implication in aging and age-related diseases. J. Clin. Invest. 2018;128:2702–2712.
Weller, J., and Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 2018 Jul 31;7:F1000 Faculty Rev-1161.
Alzheimer's disease is the most common cause of dementia worldwide, with the prevalence continuing to grow in part because of the aging world population. This neurodegenerative disease process is characterized classically by two hallmark pathologies: β-amyloid plaque deposition and neurofibrillary tangles of hyperphosphorylated tau. Diagnosis is based upon clinical presentation fulfilling several criteria as well as fluid and imaging biomarkers. Treatment is currently targeted toward symptomatic therapy, although trials are underway that aim to reduce the production and overall burden of pathology within the brain. Here, we discuss recent advances in our understanding of the clinical evaluation and treatment of Alzheimer's disease, with updates regarding clinical trials still in progress.
Wium M , Paccez J , Zerbini L . The Dual Role of TAM Receptors in Autoimmune Diseases and Cancer: An Overview. Cells. 2018;7(10):166.
Zhang M, Qian C, Zheng ZG, Qian F, Wang Y, Thu PM, et al. Jujuboside A promotes Aβ clearance and ameliorates cognitive deficiency in Alzheimer’s disease through activating Axl/HSP90/PPARγ pathway. Theranostics. 2018;8(15):4262–78.
Ambrożewicz E., Muszyńska M., Tokajuk G., Grynkiewicz G., Žarković N., Skrzydlewska E. Beneficial Effects of Vitamins K and D3 on Redox Balance of Human Osteoblasts Cultured with Hydroxypatite-Based Biomaterials. Cells. 2019;8:325.
Alisi L, Cao R, De Angelis C, Cafolla A, Caramia F, Cartocci G, et al. The relationships between vitamin K and cognition: a review of current evidence. Neurol. 2019;10:239.
In recent years, studies conducted in vitro and on animals highlighted vitamin K involvement in brain cells development and survival. In particular, vitamin K seems to have an antiapoptotic and anti-inflammatory effect mediated by the activation of Growth Arrest Specific Gene 6 and Protein S. Moreover, this vitamin is involved in sphingolipids metabolism, a class of lipids that participate in the proliferation, differentiation, and survival of brain cells. An altered expression in sphingolipids profile has been related to neuroinflammation and neurodegeneration. This review stems from a growing interest in the role of vitamin K in brain functions, especially in cognition, also in view of an expected increase of prevalence of Alzheimer's disease and other forms of dementia. Moreover, vitamin K antagonists, used worldwide as oral anticoagulants, according to recent studies may have a negative influence on cognitive domains such as visual memory, verbal fluency and brain volume. The aim of this review is to analyze the evidence of clinical studies carried out up to date on the relationship between vitamin K intake and cognitive performances.
Axmann M., Strobl W.M., Plochberger B., Stangl H. Cholesterol transfer at the plasma membrane. Atherosclerosis. 2019;290:111–117.
Cholesterol homeostasis is of central importance for life. Therefore, cells have developed a divergent set of pathways to meet their cholesterol needs. In this review, we focus on the direct transfer of cholesterol from lipoprotein particles to the cell membrane. More molecular details on the transfer of lipoprotein-derived lipids were gained by recent studies using phospholipid bilayers. While amphiphilic lipids are transferred right after contact of the lipoprotein particle with the membrane, the transfer of core lipids is restricted. Amphiphilic …
Caberlotto L., Nguyen T.P., Lauria M., Priami C., Rimondini R., Maioli S., Cedazo-Minguez A., Sita G., Morroni F., Corsi M., et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019;9:3965.
Di Daniele N, Celotto R, Fegatelli DA, Gabriele M, Rovella V, Scuteri A. Common carotid artery calcification impacts on cognitive function in older patients. High Blood Press Cardiovasc Prev. 2019 apr;26(2):127-34.
Cognitive impairment and dementia represent an emerging health problem. Cardiovascular (CV) risk factors contribute to cognitive impairment. This study investigated the effect of vascular calcification on cognitive impairment and dementia, independently of plaque and traditional CV risk factors. They found that carotid artery calcification was associated with a lower MMSE score than in subjects with no carotid calcification after controlling for age, sex, education, blood pressure levels, diabetes, creatinine, lipid lowering therapy, neuroimaging alteration, and CCA plaque. Similarly, carotid calcification was associated with higher odds of dementia regardless of the presence of CCA plaque. They concluded that CCA calcification is associated with cognitive impairment and dementia, independently of established CV risk factors and CCA plaque.
Ferland T, Allaire P, Ouliass B. Warfarin treatment alters Gas6 and Protein S activity and their downstream signaling pathways in brain, and stimulates Microglial activity (P24-002-19).Current Developments in Nutrition. 2019 June;3(S1):nzz044.P24-002-19.
There is now convincing evidence that vitamin K (VK) has important actions in the nervous system and cognition. Two VK-dependent proteins are closely linked to the brain namely Gas6 and protein S (PS). Functionally, both proteins are ligands for receptors of tyrosine kinases Tyro3, Axl, and Mer. In vitro, Gas6 and PS have been shown to possess pro-survival activity towards neurons and glia. In a previous study, targeted depletion of VK in brain induced by warfarin (W) treatment, a VK antagonist, resulted in cognitive and behavioral impairment. In the present study, we aimed to characterize the role of Gas6 and PS and their signaling pathways in W-treated rats fed or not with supplemental menaquinone-4 (MK-4), the principal K vitamer in brain. The results indicate that W treatment as used in the present study alters Gas6 and PS activity and their downstream signaling pathways, and stimulates microglial activity. Supplementing the diet with large amounts of MK-4 normalized much of the phenotype.
In this review, we will discuss the most prominent molecular mechanisms related to the PI3K-Akt pathway in AD and how T2D and altered insulin signaling may affect the pathogenesis of AD.
Guss JD, Taylor E, Rouse Z, Roubert S, Higgins CH, Thomas CJ, et al. The microbial metagenome and bone tissue composition in mice with microbiome-induced reductions in bone strength. Bone. 2019;127:146–154.
Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10:1008.
Neurodegenerative diseases share the fact that they derive from altered proteins that undergo an unfolding process followed by formation of β-structures and a pathological tendency to self-aggregate in neuronal cells. This is a characteristic of tau protein in Alzheimer's disease and several tauopathies associated with tau unfolding, α-synuclein in Parkinson's disease, and huntingtin in Huntington disease. Usually, the self-aggregation products are toxic to these cells, and toxicity spreads all over different brain areas. We have postulated that these protein unfolding events are the molecular alterations that trigger several neurodegenerative disorders. Most interestingly, these events occur as a result of neuroinflammatory cascades . In this context, the involvement of innate immunity seems to be a major paradigm in the pathogenesis of these diseases. This is an important element for the search for potential therapeutic approaches for all these brain disorders.
Herrera-Rivero M, Santarelli F, Brosseron F, Kummer MP, Heneka MT. Dysregulation of TLR5 and TAM ligands in the Alzheimer’s brain as contributors to disease progression. Mole Neurobiol. 2019 Sep;56(9):6539-6550.
The hypothesis that accumulation of beta-amyloid (Aβ) species in the brain represents a major event in Alzheimer’s disease (AD) pathogenesis still prevails; nevertheless, an array of additional pathological processes contributes to clinical presentation and disease progression. We sought to identify novel targets for AD within genes related to amyloid precursor protein (APP) processing, innate immune responses, and the catecholamine system. Through a series of bioinformatics analyses, we identified TLR5 and other genes involved in toll-like receptor (TLR) signaling as potential AD targets. Our results underscore the role of TLR dysregulations in AD, suggesting the presence of an immunosuppressive response during moderate disease stages, and implicate TAM signaling in AD immune dysregulation.
Hussain G, Wang J, Rasul Z, Anwar H, Imran A, Qasim M, et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019 Jan 25;18(1):26.
Brain is a vital organ of the human body which performs very important functions such as analysis, processing, coordination, and execution of electrical signals. For this purpose, it depends on a complex network of nerves which are ensheathed in lipids tailored myelin; an abundant source of lipids in the body. The nervous system is enriched with important classes of lipids; sphingolipids and cholesterol which compose the major portion of the brain particularly in the form of myelin. Both cholesterol and sphingolipids are embedded in the microdomains of membrane rafts and are functional units of the neuronal cell membrane. These molecules serve as the signaling molecules; hold important roles in the neuronal differentiation, synaptogenesis, and many others. Thus, their adequate provision and active metabolism are of crucial importance in the maintenance of physiological functions of brain and body of an individual. In the present review, we have highlighted the physiological roles of cholesterol and sphingolipids in the development of the nervous system as well as the association of their altered metabolism to neurological and neurodegenerative diseases.
Li M., Ye J., Zhao G., Hong G., Hu X., Cao K., Wu Y., Lu Z. Gas6 attenuates lipopolysaccharide induced TNF α expression and apoptosis in H9C2 cells through NF κB and MAPK inhibition via the Axl/PI3K/Akt pathway. Int. J. Mol. Med. 2019;44:982–994.
Lin Y-F, Smith AV, Aspelund T, Betensky RA, Smoller JW, Gudnason V, et al. Genetic overlap between vascular pathologies and Alzheimer’s dementia and potential causal mechanisms. Alzheimers Dement. 2019 Jan;15(1):65-75.
Our findings provide evidence for genetic overlap, mostly due to apolipoprotein E (APOE) gene, between vascular pathologies and AD dementia. The association between AD polygenic risk and late-life cognition is mediated in part via effects on vascular pathologies.
McCann A, Jeffery IB, Ouliass B, Ferland G, Fu X, Booth SL, Tran TTT, O'Toole PW, O'Connor EM. Exploratory analysis of covariation of microbiota-derived vitamin K and cognition in older adults. Am J Clin Nutr. 2019 Dec 1;110(6):1404-1415.
Vitamin K has multiple important physiological roles, including blood coagulation and beneficial effects on myelin integrity in the brain. Some intestinal microbes possess the genes to produce vitamin K in the form of menaquinone (MK). MK appears in higher concentration in tissues, such as the brain, particularly MK4, than the dietary form of phylloquinone (PK). Lower PK concentrations have been reported in patients with Alzheimer disease while higher serum PK concentrations have been positively associated with verbal episodic memory. The aim of the current study was to investigate the relation between genes involved in gut-microbiota derived MK, concentrations of MK isoforms, and cognitive function. Three MK isoforms were found to be positively associated with cognition, along with the identification of key components of the MK pathway that drive this association. Although the causality and direction of these associations remain unknown, these findings justify further studies. This study provides evidence that although total concentrations of MK did not covary with cognition, certain MK isoforms synthesized by the gut microbiome, particularly the longer chains, are positively associated with cognition.
Patterson W, Booth S, Dolnikowski G, Dawson-Hughes B, Morris M, Holland T, et al. Correlations of vitamin D forms and vitamin K forms within four human brain regions: The memory and aging project (MAP). Curr Dev in Nutr. 2019;3 (Supp 1): P14-025-19.
Vitamin D forms were significantly positively correlated within the four regions studied, but vitamin K forms were not. These data provide insight into a potential role for vitamin K and vitamin K in Alzheimer’s disease and dementia merits further study.
Peng CK, Wu CP, Lin JY, Peng SC, Lee CH, Huang KL. Shen CH. Gas6/Axl signaling attenuates alveolar inflammation in ischemia-reperfusion-induced acute lung injury by up-regulating SOCS3-mediated pathway. PLoS ONE. 2019;14:e0219788.
Axl is a cell surface receptor tyrosine kinase, and activation of the Axl attenuates inflammation induced by various stimuli. Growth arrest-specific 6 (Gas6) has high affinity for Axl receptor. The role of Gas6/Axl signaling in ischemia-reperfusion-induced acute lung injury (IR-ALI) has not been explored previously. They found that Gas6 attenuated IR-induced lung edema, the production of proinflammatory cytokines in perfusates, and the severity of ALI ex vivo.
Saputra WD, Aoyama N, Komai M, Shirakawa H. Menaquinone-4 suppresses lipopolysaccharide -induced inflammation in MG6 mouse microglia-derived cells by inhibiting the NF-κB signaling pathway. Int J Mol Sci. 2019;20:E2317.
Abstract: The overactivation of microglia is known to trigger inflammatory reactions in the central nervous system, which ultimately induce neuroinflammatory disorders including Alzheimer’s disease. However, increasing evidence has shown that menaquinone-4 (MK-4), a subtype of vitamin K2, can attenuate inflammation in the peripheral system. Whereas it was also observed at high levels within the brain, its function in this organ has not been well characterized. Therefore, we investigated the effect of MK-4 on microglial activation and clarified the underlying mechanism. The results indicate that MK-4 attenuates microglial inflammation by inhibiting NF-B signaling.
Spencer SJ, Basri B, Sominsky L, Soch A, Ayala MT, Reineck,P, et al. High-fat diet worsens the impact of aging on microglial function and morphology in a region-specific manner. Neurobiol. Aging. 2019; 74:121–134.
Skikantha P, Mohajeri MH. The possible role of the micobiota-gut-brain-axis in the Autism Spectrum disorder. Int J Mol Sci. 2019;20:2115.
New research points to a possible link between autism spectrum disorder (ASD) and the gut microbiota as many autistic children have co-occurring gastrointestinal problems. This review focuses on specific alterations of gut microbiota mostly observed in autistic patients. Particularly, the mechanisms through which such alterations may trigger the production of the bacterial metabolites, or leaky gut in autistic people are described. Various altered metabolite levels were observed in the blood and urine of autistic children, many of which were of bacterial origin such as short chain fatty acids (SCFAs), indoles and lipopolysaccharides (LPS). A less integrative gut-blood-barrier is abundant in autistic individuals. This explains the leakage of bacterial metabolites into the patients, triggering new body responses or an altered metabolism. Some other co-occurring symptoms such as mitochondrial dysfunction, oxidative stress in cells, altered tight junctions in the blood-brain barrier and structural changes in the cortex, hippocampus, amygdala and cerebellum were also detected. Moreover, this paper suggests that ASD is associated with an unbalanced gut microbiota (dysbiosis). Although the cause-effect relationship between ASD and gut microbiota is not yet well established, the consumption of specific probiotics may represent a side-effect free tool to re-establish gut homeostasis and promote gut health.
Rhea Em, Banks. WA. Role of the blood-brain barrier in central nervous system insulin resistance. Front Neurosci. 2019 Jun; 04 June 2019.
The blood-brain barrier (BBB) mediates the communication between the periphery and the central nervous system (CNS). Recently, CNS insulin resistance has been elucidated to play a role in neurodegenerative disease. This has stimulated a wealth of information on the molecular impact of insulin in the brain, particularly in the improvement of cognition. Since the BBB regulates the transport of insulin into the brain and thus, helps to regulate CNS levels, alterations in the BBB response to insulin could impact CNS insulin resistance. In this review, we summarize the effect of insulin on some of the cell types that make up the BBB, including endothelial cells, neurons, astrocytes, and pericytes. We also summarize how insulin can regulate levels of the pathological hallmarks of Alzheimer’s disease, including amyloid beta (Aβ) and tau within each cell type.
Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomed. 2019;14:5541–5554.
In addition to a detailed report on causative factors leading to AD, the present review also discusses the current state of the art in AD therapeutics and diagnostics, including labeling and imaging techniques employed as contrast agents for better visualization and sensing of the plaques. The review also points to an urgent need for nanotechnology as an efficient therapeutic strategy to increase the bioavailability of drugs in the central nervous system.
Tondo G, Perani D, Comi C. TAM receptor pathways at the crossroads of Neuroinflammation and neurodengeneration. Dis Markers. 2019;2019:2387614.
Increasing evidence suggests neuroinflammation is linked with neuroinflammatory responses. TAM receptors and the corresponding ligands, Growth Arrest Specific 6 and Protein S, are expressed in different tissues, including the nervous system, playing complex roles in tissue repair, inflammation and cell survival, proliferation, and migration. In the nervous system, TAM receptor signaling modulates neurogenesis and neuronal migration, synaptic plasticity, microglial activation, phagocytosis, myelination, and peripheral nerve repair, resulting in potential interest in neuroinflammatory and neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Multiple Sclerosis. In Alzheimer and Parkinson diseases, a role of TAM receptors in neuronal survival and pathological protein aggregate clearance has been suggested, while in Multiple Sclerosis TAM receptors are involved in myelination and demyelination processes. In this review, we summarise the roles of TAM receptors in the central nervous system, focusing on the regulation of immune responses and microglial activities and analysing in vitro and in vivo studies regarding TAM signalling involvement in neurodegeneration
Tran HQ, Mills RB, Peters NV, Holder MK, de Vries GJ, Knight R, et al. Associations of the fecal microbial proteome composition and proneness to diet-induced obesity. Mol Cell Proteomics. 2019 Sep;18(9):1864-1879.
Consumption of refined high-fat, low-fiber diets promotes obesity and its associated consequences. Although genetics play an important role in dictating susceptibility to such obesogenic diets, mice with nearly uniform genetics exhibit marked heterogeneity in their extent of obesity in response to such diets. This suggests non-genetic determinants play a role in diet-induced obesity. Hence, we sought to identify parameters that predict, and/or correlate with, development of obesity in response to an obesogenic diet. We assayed behavior, metabolic parameters, inflammatory markers/cytokines, microbiota composition, and the fecal metaproteome, in a cohort of mice. Fecal metaproteome analysis revealed functional and taxonomic differences among the proteins associated with proneness to obesity. Although future work will be needed to determine the breadth of applicability of these associations to other cohorts of animals and humans, this study nonetheless highlights the potential power of gut microbial proteins to predict and perhaps impact development of obesity.
Vasefi M, Hudson M, Ghaboolian-Zare E. Diet associated with inflammation and Alzheimer's Disease. J Alzheimers Dis Rep. 2019 Nov 16; 3(1):299-309.
Yuan J, Meloni BP, Shi T, Bonser A, Papadimitriou JM, Mastaglia FL, et al. The potential influence of bone-derived modulators on the progression of Alzheimer’s disease. J Alzheimers Dis. 2019;69:59-70.
Agrawal I, Jha S. Mitochondrial dysfunction and Alzheimer’s disease: role of microglia. Front Aging Neurosci. 2020;12:252.
The neurodegenerative disease known as Alzheimer’s disease (AD) is the most common cause of dementia worldwide. AD normally develops with aging and is mostly initiated because of the imbalance between the formation and clearance of amyloid-β (Aβ). Formation of neurofibrillary tangles (NFTs) of hyperphosphorylated tau is another hallmark of AD. Neuroinflammation plays a significant role in the development and pathology of AD. This chapter explores the role of mitochondrial dysfunction in microglia in case of AD. Elevated generation of reactive oxygen species (ROS) and loss of mitochondrial membrane potential through various mechanisms have been observed in AD. Elevated ROS in microglia causes activation of inflammatory and cell death pathways. Production of ATP, regulation of mitochondrial health, autophagy, and mitophagy in microglia play significant roles in the AD pathology. Understanding microglial physiology and mitochondrial dysfunction will enable better therapeutic interventions.
Alisi L, Cafolla C, Gentili A, Tartaglione S, Curini R, Cafolla A. Vitamin K concentration and cognitive status in elderly patients on anticoagulant therapy: a pilot study. J Aging Res. 2020;2020:9695324.
The study shows a positive association between vitamin K1 concentration and cognitive status in elderly patients (≥75 years) on OAT. The relationship between vitamin K1 concentration and MODA scores is described by a linear model. Cognitive status is not influenced by the duration of oral anticoagulant therapy, but by the years of education.
Annweiler G, Labriffe M, Ménager P, Ferland G, Brangier A, Annweiler C, et al. Intracranial calcifications under vitamin K antagonists or direct oral anticoagulants: results from the French VIKING study in older adults. Mauritas. 2020 Feb;132:35-39.
The use of vitamin K antagonists (VKA) is associated with the onset of vascular and soft-tissue calcifications. The objective of this study was to determine whether the regular use of VKA in older adults was associated with an increased burden of intracranial calcifications compared with the use of direct oral anticoagulant (DOA). Results: The 19 patients using VKA (median[IQR], 84years[7]; 10females) exhibited a greater burden of falcian calcifications than the 19 controls using DOA. Overall, we found that using VKA was directly associated with the global burden of intracranial calcifications. No correlation was found with calcifications in sites other than the falx cerebri. This finding may explain part of the neurocognitive morbidity met with VKA.
Chauhan A., Chauhan V. Beneficial Effects of Walnuts on Cognition and Brain Health. Nutrients. 2020;12:550.
Carrillo J.Á., Arcusa R., Zafrilla M.P., Marhuenda J. Effects of fruit and vegetable-based nutraceutical on cognitive function in a healthy population: placebo-controlled, double-blind, and randomized clinical trial. Antioxidants. 2021;10:116.
Ferland G. 2020. Vitamin K. In: Marriott BP, Birt DF, Stallings VA, Yates AA, editors. Present Knowledge in Nutrition (Eleventh Edition) Bernadette P Marriott, Diane F Birt, Allison A Yates, editors. Academic Press, Published by Elsevier Inc. p. 137-153.
Ferland G, Muhire G, Racine M-C, Ouliass B, Girouard H. Positive effects of vitamin K on arterial calcification, cerebral blood flow, and cognition in a murine model of arterial calcification. Curr Dev Nutr. 2020 Jun;4(Suppl 2):1204.
This study looked at whether VK can attenuate cognitive impairment associated with arterial stiffness. When groups were combined, MK-4 (100 mg/kg) fed mice had better cognitive abilities compared to K1 (0.1) mg/kg group. In contrast, memory retention was altered by arterial calcification in the lower VK diet groups (K1 0.1 and MK-4 10 mg/kg) (P < 0.05). MK-4 was the main form of VK in brain (at least 98.5%) irrespective of diet. Expression of carboxylated Gas6 and PS increased with dietary MK-4 but not dietary K1. The results suggest that VK can oppose the detrimental effects of vascular calcification on cognition in part by limiting vascular calcification, restoring rCBF, and increasing Gas6 and PS carboxylation in brain.
Gilchrist SE., Goudarzi S, Hafizi S. Gas6 inhibits toll-like receptor-mediated inflammatory pathways in mouse microglia via Axl and Mer. Front Cell Neurosci. 2020;14:1–10.
Microglia are well known key regulators of neuroinflammation which feature in multiple neurodegenerative disorders. These cells survey the CNS and, under inflammatory conditions, become "activated" through stimulation of toll-like receptors (TLRs), resulting in changes in morphology and production and release of cytokines. In the present study, we examined the roles of the related TAM receptors, Mer and Axl, and of their ligand, Gas6, in the regulation of microglial pro-inflammatory TNF-α production. They found Gas6 can negatively regulate the microglial pro-inflammatory response to LPS as well as via stimulation of other TLRs, acting through either of the TAM receptors, Axl and Mer. This finding indicates an interaction between TLR and TAM receptor signaling pathways and reveals an anti-inflammatory role for the TAM ligand, Gas6, which could have therapeutic potential.
Goudarzi S., Gilchrist S.E., Hafizi S. Gas6 Induces myelination through anti-inflammatory IL-10 and TGF- β upregulation in white matter and glia. Cells. 2020;9:1779.
Hadipour E, Tayarani-Najaran Z, Fereidoni M. Vitamin K2 protects PC12 cells against Aβ (1-42) and H 2 O 2-induced apoptosis via p38 MAP kinase pathway. Nutr Neurosci. 2020 May;23(5):343-52.
Alzheimer's is an age-related disease with a hallmark of progressive loss of memory formation followed by a damage in the brain function due to the neural degeneration and extracellular beta-amyloid (Aβ) plaques accumulation. This study examines the protective effects of vitamin K2 on toxicity induced by (Aβ) (1-42) and H2O2 in PC12 cells. PC12 cells pretreated with vitamin K2 (5-200 μM) for 4, 24 and 48 h, and exposed to either Aβ (for 48 h or H2O2 for 24 h. Then the protective, antioxidant and anti-apoptotic effects of vitamin K2 in PC12 cells were investigated. Vitamin K2 pretreatment (5-200 μM) significantly decreased the Aβ (1-42) and H2O2 cytotoxicity. In addition, vitamin K2 could attenuate reactive oxygen species (ROS) level after exposure of cells to H2O2 for 24 h and Aβ (1-42) for 48 h. The Aβ and H2O2 increased cell death significantly, while the pretreatment with vitamin K2 significantly reduced cell death. According to these findings, it seems that vitamin K2 possess anti-apoptotic and antioxidant effects and suggests that vitamin K2 may be a valuable protective candidate against the progression of Alzheimer's disease.
Hazan S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation : a case report. J Int Med Res. 2020;48(6):1-6.
Alzheimer’s disease (AD), the most common form of dementia, is a leading cause of death and a major cause of morbidity in older people. The disease is characterized by progressive memory loss, cognitive impairment, and the cerebral accumulation of amyloid-β peptide. Given the health and economic impacts of AD, treatments that target the underlying etiology of AD or modify the course of the disease are of significant interest. The gut microbiome has been increasingly implicated in the pathogenesis of several neurological diseases, including multiple sclerosis and Parkinson’s disease. Furthermore, emerging evidence has demonstrated that there are alterations in gut microbiome composition in patients with AD, suggesting involvement of the microbiome–gut–brain axis. We present symptom improvement in a patient with AD following fecal microbiota transplantation for a Clostridioides difficile infection.
Holsinger RMD, Elangovan S. Neuroprotective effects of fecal microbiota transplantation in a mouse model of Alzheimer’s disease. Alz & Dementia. 2020 Dec 7;6(S3).DOI:10.1002/alz.046523
The efficacy of fecal microbiota transplantation (FMT) as a therapeutic in Alzheimer's disease was evaluated in the 5XFAD mouse model.MethodWe treated sixteen, 36-week-old 5XFAD transgenic mice with fecal slurry from healthy, wild-type donors of similar age (n = 8; Old Tg-FO) or from younger (8-10-week old) healthy, wild-type donors (n = 8; Old Tg-FY) for seven days. Mice were incubated for 21 days and were then subjected to cognitive tests to examine the effects of FMT on memory. Mice were sacrificed and brain tissue was examined for amyloid plaque load.ResultImproved spatial and recognition memory in Old Tg-FY and enhanced recognition memory in Old Tg-FO mice were observed when compared to Old Tg-Control mice treated with saline. Importantly, there were significant decreases in cortical Ab loading following seven days of FMT in all treated mice.Our results demonstrate the capability FMT in improving cognition and reducing amyloid pathology in the 5XFAD mouse model of Alzheimer’s.
Kiely A, Ferland G, Ouliass B, O'Toole PW, Purtill H, O'Connor EM. Vitamin K status and inflammation are associated with cognition in older Irish adults. Nutr Neurosci. 2020:23:591–9.
The primary aim of the ELDERMET study was to investigate the relationship between gut bacteria, diet, lifestyle and health in 500 older Irish adults. Significant differences in serum phylloquinone, dietary phylloquinone and inflammatory markers were found across varying levels of cognitive function, after controlling for sex, age, body mass index (BMI), triglycerides and blood pressure. In addition, significantly higher levels of dietary phylloquinone were found in those with better cognition compared to those with the poorest function. Higher levels of inflammation were also associated with poor cognition. Furthermore, both dietary and serum phylloquinone were significant independent predictors of good cognitive function, after controlling for confounders. This study highlights the importance of dietary vitamin K as a potentially protective cognitive factor. Strategies should be devised by which elderly populations can access rich dietary sources of phylloquinone to maintain cognition.
Kim SM, DeFazio JR, Hyoju SK, Sangani K, Keskey R, Krezalek MA, et al. Fecal microbiota transplant rescues mice form human pathogen mediated sepsis by restoring systemic immunity. Nature Communications. 2020;11:2354.
Death due to sepsis remains a persistent threat to critically ill patients confined to the intensive care unit and is characterized by colonization with multi-drug-resistant healthcare-associated pathogens. Here we report that sepsis in mice caused by a defined four-member pathogen community isolated from a patient with lethal sepsis is associated with the systemic suppression of key elements of the host transcriptome that is required for pathogen clearance and decreased butyrate expression. More specifically, these pathogens directly suppress interferon regulatory factor 3. Fecal microbiota transplant (FMT) reverses the course of otherwise lethal sepsis by enhancing pathogen clearance. This protective effect is linked to the expansion of butyrate-producing Bacteroidetes. Taken together these results suggest that fecal microbiota transplantation may be a treatment option in sepsis associated with immunosuppression.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12.
Neuroinflammation is associated with neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. Microglia and astrocytes are key regulators of inflammatory responses in the central nervous system. The activation of microglia and astrocytes is heterogeneous and traditionally categorized as neurotoxic (M1-phenotype microglia and A1-phenotype astrocytes) or neuroprotective (M2-phenotype microglia and A2-phenotype astrocytes). A better understanding of the roles of microglia and astrocytes in neurodegenerative diseases is essential for developing effective therapies. In this review, we discuss the roles of inflammatory response in neurodegenerative diseases, focusing on the contributions of microglia and astrocytes and their relationship.
Machado-Fragua MD, Hoogendijk EO, Struijk EA, Rodriguez-Artalejo F, Lopez-Garcia E, Beulens JW, van Ballegooijen AJ. High dephospho-uncarboxylated matrix Gla protein concentrations, a plasma biomarker of vitamin K, in relation to frailty: the Longitudinal Aging Study Amsterdam. Eur J Nutr. 2020 Apr; 59(3):1243-1251.
No previous study has evaluated the relationship between vitamin K and frailty. Thus, we assessed the relationship between vitamin K status and frailty over 13 years in the Longitudinal Aging Study Amsterdam (LASA). They found that baseline plasma low vitamin K status was associated with a greater degree of frailty and frailty risk in this cohort of older adults, which highlights the importance of ensuring an optimal nutritional status of this vitamin to prevent frailty in later life.
Miyazawa S, Moriya S, Kokuba H, Hino H, Takano N, Miyazawa K. Vitamin K2 induces non-apoptotic cell death along with autophagosome formation in breast cancer cell lines. Breast Cancer. 2020 Mar;27(2);225-35.
Vitamin K2 (VK2) has been reported to induce apoptosis in many types of cancer cells including leukemia. However, there are no precise reports regarding the breast cancer cells. From the stand point of clinical implications of VK2 including chemoprevention, we investigated the effects of VK2 on breast cancer cell lines. They found that VK2 induced non-apoptotic cell death along with autophagy, in triple negative breast cancer cell lines. Cell death phenotype induced by VK2 appears to differ among the type of cancers. This suggests the possibility of using VK2 for the breast cancer therapy.
Moghadam BF, Fereidoni M. Neuroprotective effect of menaquinone-4 (MK-4) on transient global cerebral ischemia/reperfusion injury in rat. PLoS One. 2020;15(3):e0229769.
Cerebral ischemia/reperfusion (I/R) injury causes cognitive deficits, excitotoxicity, neuroinflammation, oxidative stress and brain edema. Vitamin K2 (Menaquinone 4, MK-4) as a potent antioxidant can be a good candidate to ameliorate I/R consequences. This study focused on the neuroprotective effects of MK-4 for cerebral I/R insult in rat’s hippocampus. The rat model of cerebral I/R was generated by transient bilateral common carotid artery occlusion for 20 min. Rats were divided into control, I/R, I/R+DMSO (solvent (1% v/v)) and I/R+MK-4 treated (400 mg/kg, i.p.) groups. Twenty-four hours after I/R injury induction, total brain water content, superoxide dismutase (SOD) activity, nitrate/nitrite concentration and neuronal density were evaluated. The findings indicated that administration of MK-4 following I/R injury improved anxiety-like behavior, short term and spatial learning and memory impairment induced by I/R. Also, MK-4 was able to diminish the increased total brain water content, apoptotic cell density, Bax/ Bcl2 ratio and GFAP mRNA expression following I/R. In addition, the high level of nitrate/nitrite, IL-6, IL-1β and TNF-α induced by I/R was reduced after MK-4 administration. The findings demonstrated that MK-4 can rescue transient global cerebral I/R consequences via its anti-inflammatory and anti-oxidative stress features. MK-4 administration ameliorates neuroinflammation, neurotoxicity and neuronal cell death processes and leads to neuroprotection. This study highlights that very high levels of MK4 available in the brain greatly improved healing from an stroke.
Nguyen TT, Ta QTH, Nguyen TTD, Le TT, Vo VG. Role of insulin resistance in the Alzheimer’s disease Ppogression. Neurochem. Res. 2020 doi: 10.1007/s11064-020-03031-0.
Insulin resistance or dysfunction of insulin signaling is a universal feature of T2D, the main culprit for altered glucose metabolism and its interdependence on cell death pathways, forming the basis of linking T2D with AD as it may exacerbate Aβ accumulation, tau hyperphosphorylation and devastates glucose transportation, energy metabolism, hippocampal framework and promulgate inflammatory pathways. The current work demonstrates the basic mechanisms of the insulin resistance mediates dysregulation of bioenergetics and progress to AD as a mechanistic link between diabetes mellitus and AD.
Pant DC, Aguilera-Albesa S, Pujol A. Ceramide signalling in inherited and multifactorial brain metabolic diseases. Neurobio of Dis. 2020 Sept;143:105014.
In the present review, we highlight the critical role of ceramide signaling in several different neurological disorders as well as the effects of their perturbations and discuss how this emerging class of bioactive sphingolipids has attracted interest in the field of neurological diseases.
Quinn L, Sheh A, Ellis JL, Smith DE, Booth SL, Fu X, Muthupalani S, et al. Helicobacter Pylori antibiotic eradication coupled with a chemically defined diet in INS-GAS Mmce triggers dysbiosis and Vvtamin K deficiency resulting in gastric hemorrhage. Gut Microbes. 2020;11:820–841.
To determine potential synergistic effects, H. pylori-infected male INS-GAS mice were fed an amino acid defined (AAD) diet with increased folate and were treated with antibiotics after 18 weeks of H. pylori infection. Antibiotic therapy decreased gastric pathology, but dietary folate had no effect. However, the combination of antibiotics and the AAD diet induced anemia, gastric hemorrhage, and mortality. Clinical presentation suggested hypovitaminosis K potentially caused by dietary deficiency and dysbiosis. Based on current dietary guidelines, the AAD diet was deficient in vitamin K. Phylloquinone administered subcutaneously and via a reformulated diet led to clinical improvement with no subsequent mortalities and increased hepatic vitamin K levels. We characterized the microbiome and menaquinone profiles of antibiotic-treated and antibiotic-free mice. Antibiotic treatment decreased the abundance of menaquinone producers within orders Bacteroidales and Verrucomicrobiales. PICRUSt predicted decreases in canonical menaquinone biosynthesis genes, menA and menD. Reduction of menA from Akkermansia muciniphila, Bacteroides uniformis, and Muribaculum intestinale were confirmed in antibiotic-treated mice. The fecal menaquinone profile of antibiotic-treated mice had reduced MK5 and MK6 and increased MK7 and MK11 compared to antibiotic-free mice. Loss of menaquinone-producing microbes due to antibiotics altered the enteric production of vitamin K. This study highlights the role of diet and the microbiome in maintaining vitamin K homeostasis.
Rusu IG, Suharoschi R, Vodnar DC, Pop CR. Socaci SA, Vulturar R, et al. Iron supplementation influence on the gut microbiota and probiotic intake effect in iron deficiency—A literature-based review. Nutrients. 2020;12:1993.
Sanada J, Nakajima S, Kurokawa S, Barelo-Soler A, Ikuse D, Hirata A, et al. Gut microbiota and major depressive disorder: a systematic review and meta-analysis. J Affect Disord. 2020 apr 1;266:1-13.
Growing attention has been paid to the field of gut microbiota for mental disorders over the last decade. However, to our knowledge, no studies have conducted systematic reviews on the association between gut microbiota and major depressive disorder (MDD) in both interventional and non-interventional studies. Our results indicate that several taxa at the family and genus levels, specifically family Prevotellaceae, genus Corprococcus, and Faecalibacterium, were decreased in MDD compared to non-depressed controls in observational studies, and depressive symptoms were improved compared to controls in interventional studies with probiotics. Due to the limited number of studies, further studies considering diet and pharmacotherapy are needed to explore the relationships between gut microbiota and MDD in humans.
Tanprasertsuk J, Ferland G, Johnson MA, Poon LW, Scott TM, Barbey AK, Barger K, Wang XD, Johnson EJ. Concentrations of circulating phylloquinone, but not cerebral menaquinone-4, are positively correlated with a wide range of cognitive measures: Exploratory Findings in Centenarians J Nutr. 2020 Jan 1; 150(1):82-90.
Roles of VK on cognitive health in the elderly are emerging. The primary objective of this study was to characterize VK distribution in brains of an elderly population with varied cognitive function. In addition, associations among circulating (a biomarker of VK intake) and cerebral VK concentrations and cognition were investigated. MK-4 was the predominant vitamer in both frontal cortex and temporal cortex, regardless of cognitive status. Antithrombotic users had 34.0% and 53.9% lower MK-4 concentrations in FC (P < 0.05) and TC (P < 0.001), respectively. Circulating PK was not correlated with cerebral MK-4 or total VK concentrations. Circulating PK concentrations were significantly associated with a wide range of cognitive measures in nondemented centenarians (P < 0.05). In contrast, cerebral MK-4 concentrations were not associated with cognitive performance, either before or after exclusion of antithrombotic users. Circulating PK may reflect intake of VK-rich foods containing other dietary components beneficial to cognitive health. Further investigation of VK uptake and metabolism in the brain is warranted.
Yang R.-Y, Pan J-Y., Chen Y, Li Y, Wu J, Wang X.-D. Menaquinone-7 protects astrocytes by regulating mitochondrial function and inflammatory response under hypoxic conditions. Eur. Rev. Med. Pharmacol. Sci. 2020;24:10181–10193.
Our study is the first to confirm that MK-7 can protect astrocytes from hypoxia-induced cytotoxicity, possibly by inhibiting mitochondrial dysfunction and the expression of proinflammatory cytokines. Gas6 may also participate in these protective effects.
Yu Z, Ling Z, Lu L, Zhao J, Chen X, Xu P, et al. Regulatory roles of bone in neurodengenerative diseases. Front Aging Neurosci. 2020;12:610581.
Osteoporosis and neurodegenerative diseases are two kinds of common disorders of the elderly, which often co-occur. Previous studies have shown the skeletal and central nervous systems are closely related to pathophysiology. As the main structural scaffold of the body, the bone is also a reservoir for stem cells, a primary lymphoid organ, and an important endocrine organ. It can interact with the brain through various bone-derived cells, mostly the mesenchymal and hematopoietic stem cells (HSCs). The bone marrow is also a place for generating immune cells, which could greatly influence brain functions. Finally, the proteins secreted by bones (osteokines) also play important roles in the growth and function of the brain. This article reviews the latest research studying the impact of bone-derived cells, bone-controlled immune system, and bone-secreted proteins on the brain, and evaluates how these factors are implicated in the progress of neurodegenerative diseases and their potential use in the diagnosis and treatment of these diseases.
Burstyn-Cohen T, Quan N. TAM signaling in the nervous system. Brain Plast. 2021;7(1):33-46.
Tyro3, Axl and Mertk are members of the TAM family of tyrosine kinase receptors. TAMs are activated by two structurally homologous ligands GAS6 and PROS1. TAM receptors and ligands are widely distributed and often co-expressed in the same cells allowing diverse functions across many systems including the immune, reproductive, vascular, and the developing as well as adult nervous systems. This review will focus specifically on TAM signaling in the nervous system, highlighting the essential roles this pathway fulfills in maintaining cell survival and homeostasis, cellular functions such as phagocytosis, immunity and tissue repair. Dysfunctional TAM signaling can cause complications in development, disruptions in homeostasis which can rouse autoimmunity, neuroinflammation and neurodegeneration. The development of therapeutics modulating TAM activities in the nervous system has great prospects, however, foremost we need a complete understanding of TAM signaling pathways.
Chen H, Shang D, Wen Y, Liang C. Bone-derived modulators that regulate brain function: emerging therapeutic targets for neurological disorders. Front Cell Dev Biol. 2021;9:683457.
This review summarizes current findings regarding the roles of these bone-derived modulators in the brain and also follows their involvement in the pathogenesis of neurological disorders. This review may aide in the development of promising therapeutic strategies for neurological disorders via targeting bone.
De Oliveira L.G, Angelo YDS, Iglesias AH, Peron JPS. Unraveling the Link between mitochondrial dynamics and neuroinflammation. Front Immunol. 2021;12:624919.
Thus, although previously solely seen as power suppliers to organelles and molecular processes, it is now well established that mitochondria have many other important roles, including during immune responses. Here, we discuss the importance of these mitochondrial dynamics during neuroinflammation, and how they correlate either with the amelioration or worsening of CNS disease.
Gilchrist SE, Pennelli GM, Hafizi S. Gas6/TAM Signalling Negatively Regulates Inflammatory Induction of GM-CSF in Mouse Brain Microglia. Cells. 2021;10(12):3281.
Microglia and astrocytes are the main CNS glial cells responsible for the neuroinflammatory response, where they release a plethora of cytokines into the CNS inflammatory milieu. The TAM (Tyro3, Axl, Mer) receptors and their main ligand Gas6 are regulators of this response, however, the underlying mechanisms remain to be determined. We investigated the ability of Gas6 to modulate the CNS glial inflammatory response to lipopolysaccharide (LPS), a strong pro-inflammatory agent, through a qPCR array that explored Toll-like receptor signalling pathway-associated genes. The results illustrate microglia as a major resident CNS cellular source of GM-CSF as part of the neuroinflammatory response, and that Gas6/TAM signalling inhibits this response through suppression of NF-κB signalling.
Garad M., Edelmann E., Leßmann V. Impairment of spike-timing-dependent plasticity at Schaffer Collateral-CA1 synapses in adult APP/PS1 mice depends on proximity of Aβ plaques. Int J Mol Sci. 2021;22:1378.
Huang S-H, Fang S-T, Chen Y-C. Molecular mechanism of vitamin K2 protection against Amyloid-β-Induced cytotoxicity. Biomolecules. 2021;11:423.
The pathological role of vitamin K2 in Alzheimer’s disease (AD) involves a definite link between impaired cognitive functions and decreased serum vitamin K levels. Vitamin K2 supplementation may have a protective effect on AD. However, the mechanism underlying vitamin K2 protection has not been elucidated. With the amyloid-β (Aβ) cascade hypothesis, we constructed a clone to study the protective effect of vitamin K2 against Aβ cytotoxicity. They found that vitamin K2 treatment dose-dependently decreased the death of neural cells. The protective effect of vitamin K2 could be abolished by adding warfarin, a vitamin K2 antagonist. The addition of vitamin K2 reduced the ROS formation and inhibited the apoptosis induced by Aβ peptides, indicating that the mechanism underlying the vitamin K2 protection is likely against Aβ-mediated apoptosis. Our study demonstrates that vitamin K2 can protect neural cells against Aβ toxicity.
Huang Y, Happonen KE, Burrola PG, O’Connor C, Hah N, Huang L, et al. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat Immunol. 2021 May;22(5:586-594.
Two microglial TAM receptor tyrosine kinases - Axl and Mer - have been linked to Alzheimer’s disease, but their roles in disease have not been tested experimentally. We find that in Alzheimer’s disease and its mouse models, induced expression of Axl and Mer in amyloid plaque-associated microglia was coupled to induced plaque decoration by the TAM ligand Gas6 and its co-ligand phosphatidylserine. Genetic ablation of Axl and Mer resulted in microglia that were unable to normally detect, respond to, organize, or phagocytose amyloid beta plaques. These major deficits notwithstanding, TAM-deficient APP/PS1 mice developed fewer dense-core plaques than APP/PS1 mice with normal microglia. Our findings reveal that the TAM system is an essential mediator of microglial recognition and engulfment of amyloid plaques, and that TAM-driven microglial phagocytosis does not inhibit, but rather promotes, dense-core plaque development.
Lai Y, Masatoshi H, Ma Y, Guo Y, Zhang B. Role of vitamin K in intestinal health. Front Immunol. 2021;12:791565.
Intestinal diseases, such as inflammatory bowel diseases (IBDs) and colorectal cancer (CRC) generally characterized by clinical symptoms, including malabsorption, intestinal dysfunction, injury, and microbiome imbalance, as well as certain secondary intestinal disease complications, continue to be serious public health problems worldwide. The role of vitamin K (VK) on intestinal health has drawn growing interest in recent years. Several investigations continue to explore the role of VK as an emerging novel biological compound with the potential function of improving intestinal health. This study aims to present a thorough review on the bacterial sources, intestinal absorption, uptake of VK, and VK deficiency in patients with intestinal diseases, with emphasis on the effect of VK supplementation on immunity, anti-inflammation, intestinal microbes and its metabolites, antioxidation, and coagulation, and promoting epithelial development. Besides, VK-dependent proteins (VKDPs) are another crucial mechanism for VK to exert a gastroprotection role for their functions of anti-inflammation, immunomodulation, and anti-tumorigenesis. In summary, published studies preliminarily show that VK presents a beneficial effect on intestinal health and may be used as a therapeutic drug to prevent/treat intestinal diseases, but the specific mechanism of VK in intestinal health has yet to be elucidated.
Lin X, Wen X, Wei Z, Guo K, Shi F, Huang T, et al. Vitamin K2 protects against Aβ42-induced neurotoxicity by activating autophagy and improving mitochondrial function in Drosophilia. Neuroreport. 2021 Apr 7; 32(6):431-37.
Alzheimer disease is characterized by progressive decline in cognitive function due to neurodegeneration induced by accumulation of Aβ and hyperphosphorylated tau protein. This study was conducted to explore the protective effect of vitamin K2 against Aβ42-induced neurotoxicity. Alzheimer disease flies were treated with vitamin K2 for 28 days after eclosion. They found that vitamin K2 improved climbing ability, prolonged lifespan and decreased Aβ42 levels, upregulated the expression of LC3 and Beclin1, increased the conversion of LC3I to LC3II and decreased p62 level in Alzheimer disease flies. In addition, vitamin K2 upregulated the expression of NDUFS3 (P = 0.001) and increased ATP production (P = 0.0033) in Alzheimer disease flies. They concluded that vitamin K2 protect against Aβ42-induced neurotoxicity by activation of autophagy and rescue mitochondrial dysfunction, which suggests that it may be a potential valuable therapeutic approach for Alzheimer disease.
Maresz K. Growing evidence of a proven mechanism shows vitamin K2 can impact health conditions beyond bone and cardiovascular. Integr Med (Encinitas). 2021 Aug;20(4):34-38.
Vitamin K2 is a vital nutrient newly recognized for supporting bone and cardiovascular health, shown in observational and intervention trials, in healthy and patient populations, in adults and children. Even more recently, it has come to light that K2 status and the vitamin’s mechanism of action impact other health areas, including but not limited to brain health, healthy joints, neuropathy, and vision health. This evidence lends itself to the argument that correcting a widespread vitamin K2 deficiency can significantly improve global health. The first step in remedying that deficiency is establishing a vitamin K2-specific recommended daily intake.
Melzer TM, Manosso LM, Yau S-U, Gil-Mohapel J, Brocardo PS. In pursuit of healthy aging: effects of nutrition on brain function. Int J Mol Sci. 2021 May;22(9):5026.
Consuming a balanced, nutritious diet is important for maintaining health, especially as individuals age. Several studies suggest that consuming a diet rich in antioxidants and anti-inflammatory components such as those found in fruits, nuts, vegetables, and fish may reduce age-related cognitive decline and the risk of developing various neurodegenerative diseases. Numerous studies have been published over the last decade focusing on nutrition and how this impacts health. The main objective of the current article is to review the data linking the role of diet and nutrition with aging and age-related cognitive decline. Specifically, we discuss the roles of micronutrients and macronutrients and provide an overview of how the gut microbiota-gut-brain axis and nutrition impact brain function in general and cognitive processes in particular during aging. We propose that dietary interventions designed to optimize the levels of macro and micronutrients and maximize the functioning of the microbiota-gut-brain axis can be of therapeutic value for improving cognitive functioning, particularly during aging.
NIA-Funded Active Alzheimer’s and Related Dementias Clinical Trials and Studies. (accessed on 27 May 2021); Available online: www.nia.nih.gov/research/ongoing-AD-trials.
Popa DS. The role of vitamin K in humans; implication in aging and age-associated diseases. Antioxidants. 2021;10(4):566.
As human life expectancy is rising, the incidence of age-associated diseases will also increase. Scientific evidence has revealed that healthy diets, including good fats, vitamins, minerals, or polyphenolics, could have antioxidant and anti-inflammatory activities, with antiaging effects. Recent studies demonstrated that vitamin K is a vital cofactor in activating several proteins, which act against age-related syndromes. Thus, vitamin K can carboxylate osteocalcin (a protein capable of transporting and fixing calcium in bone), activate matrix Gla protein (an inhibitor of vascular calcification and cardiovascular events) and carboxylate Gas6 protein (involved in brain physiology and a cognitive decline and neurodegenerative disease inhibitor). By improving insulin sensitivity, vitamin K lowers diabetes risk. It also exerts antiproliferative, proapoptotic, autophagic effects and has been associated with a reduced risk of cancer. Recent research shows that protein S, another vitamin K-dependent protein, can prevent the cytokine storm observed in COVID-19 cases. The reduced activation of protein S due to the pneumonia-induced vitamin K depletion was correlated with higher thrombogenicity and possibly fatal outcomes in COVID-19 patients. Our review aimed to present the latest scientific evidence about vitamin K and its role in preventing age-associated diseases and/or improving the effectiveness of medical treatments in mature adults ˃50 years old.
Popescu A, German M. Vitamin K2 holds promise for Alzheimer’s prevention and treatment. Nutrients. 2021 Jun 27;13(7):2206.
Recent studies have highlighted the importance of vitamin K2 (VK2) in human health. However, there have been no clinical studies investigating the role of VK2 in the prevention or treatment of Alzheimer's disease (AD). In reviewing basic science research and clinical studies that have connected VK2 to factors involved in AD pathogenesis, we have found a growing body of evidence demonstrating that VK2 has the potential to slow the progression of AD and contribute to its prevention. In our review, we consider the antiapoptotic and antioxidant effects of VK2 and its impact on neuroinflammation, mitochondrial dysfunction, cognition, cardiovascular health, and comorbidities in AD. We also examine the link between dysbiosis and VK2 in the context of the microbiome's role in AD pathogenesis. Our review is the first to consider the physiological roles of VK2 in the context of AD, and, given the recent shift in AD research toward nonpharmacological interventions, our findings emphasize the timeliness and need for clinical studies involving VK2.
Qiu S, Palavicini JP, Wang J, Gonzalez NS, He S, Dustin E, et al. Adult-onset CNS myelinsulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Molecular Neurodegeneration. 2021;16:64.
Human genetic association studies point to immune response and lipid metabolism, in addition to amyloid-beta (Aβ) and tau, as major pathways in Alzheimer’s disease (AD) etiology. Accumulating evidence suggests that chronic neuroinflammation. Our group and others have reported early and dramatic losses of brain sulfatide in AD cases and animal models that are mediated by ApoE. They found that mild central nervous system (CNS) sulfatide losses within myelinating cells are sufficient to activate disease-associated microglia and astrocytes, and to increase the expression of AD risk genes (e.g., Apoe, Trem2, Cd33, and Mmp12), as well as previously established causal regulators of the immune/microglia network in late-onset AD. Notably, neuroinflammation and mild cognitive impairment showed gender differences, being more pronounced in females than males. Our results strongly suggest that sulfatide deficiency is an important contributor and driver of neuroinflammation and mild cognitive impairment in AD pathology.
Shandilya S, Kesari KK, Ruokolainen J. Vitamin K2 modulates organelle damage and tauopathy induced by streptozotocin and menadione in SH-SY5Y cells. Antioxidants (Basel). 2021 Jun 20;10(6):983.
Vitamin K2, known for its antioxidative and anti-inflammatory properties, can act as a potent neuroprotective molecule. To understand the neuroprotective effect of vitamin K2 during metabolic complications, SH-SY5Y cells were treated with streptozotocin for 24 h and menadione for 2 h in a dose-dependent manner, followed by post-treatment of vitamin K2 for 5 hours. Results showed that vitamin K2 significantly reduces neuronal cell death by inhibiting cytotoxicity and ROS levels and helps in the retainment of mitochondrial membrane potential. These results suggested that vitamin K2 alleviated mitochondrial damage, ER stress and tauopathy-mediated neuronal cell death, which highlights its role as new antioxidative therapeutics targeting related cellular processes.
Wilson EN, Andreasson KI. TAM-ping down Amyloid in Alzheimer’s Disease. Nat Immunol. 2021 May;22(5):543-544.
The TAM receptor kinases Axl and Mer are critical for microglial recognition and clearance of accumulating amyloid in transgenic models of Alzheimer’s disease.
Xia C, Vonder M, Sidorenkow G, Ma R, Oudkerk M, van der Harst P, et al. Coronary artery calcium and cognitive function in Dutch adults: cross-sectional results of the population-based ImaLife study. J Am Heart Assoc. 2021 Feb 16;10(4):e018172.
The aim of this study was to investigate whether increased severity of coronary artery calcium (CAC), an imaging biomarker of subclinical coronary atherosclerosis, is associated with worse cognitive function independent of cardiovascular risk factors in a large population-based Dutch cohort with broad age range. They found in this Dutch population of ≥45 years, increased CAC severity was associated with worse performance of working memory, independent of classical cardiovascular risk factors. The inverse relationship of CAC score categories with working memory was strongest in participants aged 45 to 54 years.
Azuma K, Osuka Y, Kojima N, Sasai H, Kim H, Inoue S. Association of vitamin K insufficiency with cognitive dysfunction in community-dwelling older adults. Front Nutr. 2022 Jan 31;8:811831.
Vitamin K is a fat-soluble vitamin shown to be associated with several age-related diseases. Here, we demonstrated the association of the concentration of undercarboxylated osteocalcin (ucOC) in serum, which is a biomarker for vitamin K insufficiency, with cognitive function in a cross-sectional study. A total of 800 community-dwelling older adults (mean age = 75.9) were invited to geriatric health examination. We found a significant association of impaired cognitive function and concentration of ucOC in the highest tertile of ucOC. When the analysis was repeated with each domain of cognitive measurement, the highest tertile of ucOC was associated with impaired orientation, calculation, and language. As far as we know, this is the first report on the significant association of single ucOC measurement and cognitive impairment. Our analysis also suggests that vitamin K insufficiency could be associated with selected categories of cognitive function. Since the single measurement of ucOC in serum is a simple and widely available method for vitamin K evaluation, it could be useful as a biomarker of neurodegenerative diseases affecting the cognitive functions.
Carrillo JÁ, Arcusa R, Zafrilla MP, Marhuenda J. Effects of fruit and vegetable-based nutraceutical on cognitive function in a healthy population: placebo-controlled, double-blind, and randomized clinical trial. Antioxidants. 2021;10:116.
Booth SL, Shea MK, Barger K, Leurgans SE, James BD, Holland TM, et al. Association of vitamin K with cognitive decline and neuropathology in community-dwelling older persons. Alzheimer’s Dement. 2022;8:312255.
Elkattawy HA, Ghoneim FM, Eladl MA, Said E, Ebrahim HA, El-Shafey M, et al. Vitamin K2 (Menaquinone-7) revereses age-related structural and cognitive deterioration in naturally aged rats. Antioxidants. 2022;11:514.
Aging is a naturally occurring process inevitably affecting each living human. The brain is adversely affected by aging with increased risks of developing various neurological disorders. Thus, it is essential to investigate practical approaches that can counteract the impact of aging on the brain. Vitamin K2 (MK7) is a naturally occurring vitamin with reported valuable therapeutic effects. The current study highlights the role of MK7 administration in counteracting age-related changes in the brain using naturally aging rats. Three-month-old rats were assigned to two groups: an ageing control group receiving a drug vehicle and an ageing group were given very large doses of MK7(30 mg/kg, once daily 5 days per week). Treatment was continued for 17 months. MK7 improved functional performance, reduced social anxiety, depressive-like behavior, and enhanced memory performance with concomitant preservation of hippocampal and cerebral cortex tyrosine hydroxylase expression. Biochemically, Vit. K2 administration restored oxidative-anti-oxidative homeostasis in the brain. MK7 modulated inflammatory signaling, as evidenced by suppression in the brain of NLRP3, caspase-1, Il-1β, TNFα, IL-6, and CD68 expression. Concomitantly, histopathological examination revealed consistent hippocampal and cerebral cortex improvement. Thus, it can be inferred that Vit K2 can slow down age-related changes in the brain.
NIA-Funded Active Alzheimer’s and Related Dementias Clinical Trials and Studies. [(accessed on 27 April 2022)]; Available online: www.nia.nih.gov/research/ongoing-AD-trials.
Shearer M. The biosynthesis of menaquinone-4: how a historic biochemical pathway Is changing our understanding of vitamin K nutrition. J Nutr. 2022;152(4):917-919.
2021 Alzheimer’s Disease Facts and Figures. Available online: www.alz:media/documents/alzheimers-facts-and-figures.pdf (accessed on 27 May 2021).
Tang H, Zheng Z, Wang H, Wang L, Zhao G, Wang P. Vitamin K2 modulates mitochondrial dysfunction induced by 6-hydroxydopamine in SH-SY5Y cells via mitochondrial quality-control loop. Nutrients. 2022 Apr;14(7):1504.
Vitamin K2, a natural fat-soluble vitamin, is a potent neuroprotective molecule, owing to its antioxidant effect, but its mechanism has not been fully elucidated. Therefore, we stimulated SH-SY5Y cells with 6-hydroxydopamine (6-OHDA) in a proper dose-dependent manner, followed by a treatment of vitamin K2. They found that that vitamin K2 can release mitochondrial damage, and that this effect is related to the participation of vitamin K2 in the regulation of the mitochondrial quality-control loop, through the maintenance of the mitochondrial quality-control system, and repair mitochondrial dysfunction, thereby alleviating neuronal cell death mediated by mitochondrial damage.
Wang A, Zhao M, Luo J, Zhang T, Zhang D. Association of dietary vitamin K intake with cognition in the elderly. Front Nutr. 2022;9:900887. Doi:10.3389/fnut.2022.900887.
This study examined the correlation between dietary vitamin K intake and the cognition of the elderly, using NHANES data. They found that the level of dietary vitamin K intake was inversely related to the risk of low cognitive performance in the elderly, meaning if your intake was low, you had a higher risk of reduced cognitive levels.
Coppens S, Lehmann S, Hopley C, Hirtz C. Neurofilament-Light, a promising biomarker: analytical, metrological and clinical challenges. Int J Mol Sci. 2023 Jul 19;24(14):11624. Doi: 10.339/ijms241411624
Cronje HT, Liu X, Odden MC, Moseholm KF, Seshardri S Satizabal CL. Serum NfL and GFAP are associated with incident dementia and dementia mortality in older adults, t: the cariovascular health study. Alzheimers Dement. 2023;19:5672-80.
Luo J, Lin S. Dietary vitamin K intake is associated with decreased neurofilament light chain among middle-aged and older adults from the NHANES. Front Nutr. 2024 Sep 13;11:1396707. Doi:10.3389/fnut.2024.1396707
"In life, in any given situation, one can either be a winner, a loser, or a victim.
If you keep your nose clean, work hard, work smart, and pay attention,
you will win far more than you lose - - - but every now and then you will
lose - - - no worries - - - but what is unacceptable is being a victim.
I chose not be a victim.” - Pat
Click here to read his story
Koncentrated K
USA
Phone: (715) 612-5858
Copyright © 2025 Koncentrated K, web content in part or in whole may not be used without written consent. All rights reserved.















 Web Design: www.superiorweb.net
Web Design: www.superiorweb.net